
Abstract
Background:
The advent of three effective disease modifying therapies for SMA has highlighted the need to understand the epidemiology of spinal muscular atrophy (SMA) and its disability impact.
Objective:
We aimed to establish the nationwide incidence and prevalence of SMA in Aotearoa-New Zealand, and to estimate the patients’ disability and the impact of this on health resource utilisation.
Methods:
We used multiple sources to identify patients with SMA and verified the diagnosis, disabilities and resources utilisation by review of the individual patient notes and genetic results. The four year incidence period was from 1st July 2015 to 30th June 2019. Prevalence date was 1st March 2019. Of note, this time period pre-dated access to disease modifying therapy in New Zealand. Census data for 2018 was used for denominators. Descriptive statistics and capture-recapture were used to analyse the data. For context, we reviewed international SMA epidemiology.
Results:
The incidence per 100,000 live births was 8.0 (95% confidence interval (CI): 4.8–12.5). The standardised prevalence rate of SMA on 1st March 2019 was 1.78 per 100,000 (95% CI: 1.24, 2.33). Prevalence was significantly lower amongst Māori at 0.34 (95% CI: 0.08, 1.13; p = 0.006). Substantial decline from best motor milestone performance was seen; seven patients with SMA1 died without access to disease modifying therapy. 74% of the total cohort used wheelchairs. 23% required respiratory support. 62% had scoliosis, of whom 61% had had surgery. Surviving SMA1 patients had very high health service utilisation.
Conclusions:
Incidence and prevalence figures match closely with international studies. This is the first record of low SMA rates in Māori. While the largest burden of disease falls on patients with SMA1 and 2 there is still substantial use of health resources among SMA3 and SMA4 patients.
Introduction
We set out to investigate the epidemiology of spinal muscular atrophy (SMA) in the whole country of Aotearoa-New Zealand (population 2018, 5.02 million people). The advent of three effective disease modifying therapies for SMA (nusinersen, 1 risdiplam 2 and onasemnogene abeparvovec 3 ) has made epidemiological research imperative for treatment planning. Spinal muscular atrophy (SMA) refers to a group of autosomal recessive neuromuscular diseases which are characterised by progressive degeneration of alpha motor neurones in the brainstem and spinal cord.4,5 Most cases are caused by homozygous deletion in the survival motor neurone 1 gene (SMN1) on chromosome 5q though some patients are compound heterozygous for deletion and a pathogenic variant. 6 Affected patients are largely dependent on their neighbouring SMN2 gene(s) for SMN protein production. As individuals have different numbers of SMN2 genes the disease severity differs with those having a greater number of SMN2 gene copies having milder disease. 7 This leads to a classification of SMA subtypes 0–4 based on the age of onset and the best motor milestone reached 8 (Table 1). At the time of the study none of these treatments were government funded in New Zealand so in some cases families moved overseas to access funded treatment. We studied the distribution of SMA subtypes, the age of patients, their disability and use of services, their geographical distribution and their ethnicity. While the incidence of SMA type 1 is higher than the other subtypes, SMA1 is less prevalent because of its short life expectancy without treatment. Therefore, we undertook both an incidence and a prevalence study.
SMA type definitions.

Classification of SMA types is based on age at onset and maximum motor function.
An important aspect of the condition, especially when considering the cost of expensive disease modifying medical therapies is the alternative cost of treating the condition symptomatically, so we also looked into use of health resources in our medically untreated population.
In studying population figures for Aotearoa-New Zealand it is important to understand the underlying population structure. 16.5% of the population identifies as Māori – the country's indigenous people (https://www.stats.govt.nz/topics/census#2018-census). A further 8.1% identify as being from Pacific countries with the largest numbers being from Fiji, Samoa, Tonga and the Cook Islands 9 with a relatively young demographic. “Asian” people make up 15.1% and people of European descent make up 70.2%. Total percentages exceed 100 percent because of the convention in the New Zealand census – which we used for our denominator - to allow individuals to choose more than one ethnicity (Ministry of Health. 2017. HISO 10001:2017 Ethnicity Data Protocols. Wellington: Ministry of Health).
Materials and methods
Case ascertainment
Our retrospective study of SMA prevalence and incidence collected data from multiple sources: Pūnaha Io - the NZ Neuro-Genetic Registry & BioBank (formerly the NZ NMD Registry) a voluntary registry open to patients who want to take part in research; 10 patient support organisations including the Muscular Dystrophy Association of NZ; Genetic Health Services NZ; the three clinical genetics laboratories in New Zealand; Ministry of Health national disability service Needs Assessment Service Coordination (NASC) database; hospital records and self/family referrals. In addition, all nine paediatric neurologists and the 63 adult neurologists, practising either in private or public during the study period, were contacted directly regarding known diagnoses. Responses were received from all nine paediatric and 60 adult neurologists.
Data collection started in June 2019 so we set a prevalence date of 1st March 2019 and the four-year period, 1st July 2015–30th June 2019 was selected as the incidence period as this was thought to be a long enough period to accommodate natural year-to-year variation in incidence.
Disability and health resource utilisation data
Permission from the New Zealand Health and Disability Ethics Committee (#19/NTA/37) allowed public health system medical records to be obtained and reviewed without requiring patient consent, to avoid bias which might be introduced if patients could opt out. Medical records for all identified participants were requested via the Clinical Records Departments of the New Zealand public health system. In some areas these were available electronically. In other areas, and for older members of the cohort, paper files were retrieved. In New Zealand, all children with SMA are treated by public health services. Starship Children's Hospital in Auckland is a nationwide quaternary referral centre. There is no alternative private paediatric hospital service.
All medical files pertaining to the individuals’ lifespan were reviewed by one member of the research team. The following information was collected: Age, gender, ethnicity (self-identified during any health-related consultation), and geographical region at diagnosis and point prevalence date. Diagnosis and SMA subtype were confirmed by reviewing relevant medical records and genetic test results. Patients who had a positive genetic test (either homozygous 5q deletion or compound deletion and point mutation) or who had the same phenotype as a sibling with a positive genetic test were included. SMN2 copy number was collected when available, but has not been routinely tested prior to availability of disease modifying therapies. Clinical information collected from the medical record included: age at diagnosis, maximum motor milestone, current motor ability, wheelchair use, nasogastric tube or gastrostomy insertion, respiratory support (cough assist, daytime non-invasive ventilation and/or nocturnal non-invasive ventilation), scoliosis, scoliosis surgery, medications, and treatment with disease modifying therapy. The total number of inpatient hospital visits, ED visits, outpatient visits (including allied health team contacts) and operating theatre visits were counted from medical records.
Statistical analysis
Descriptive statistics were used to characterise the sample with numbers and percentages, means and standard deviations, or medians and interquartile ranges used as appropriate. Stata software (version 16) was used for all analysis. 11 For prevalence denominators, population estimates were taken from the census data at 30 June 2018 (https://www.stats.govt.nz/topics/census#2018-census) to allow calculations by age, sex, region and ethnic group. Following the methodology of previous studies, 12 the denominator for incidence in each 12 months of the study was the total number of live births in NZ at of the mid-point of that period (https://infoshare.stats.govt.nz/). Incidence rates were calculated overall and by study year, SMA subtype and ethnic group. The Poisson distribution was used to calculate exact 95% confidence intervals for crude prevalence and incidence rates. The overall SMA prevalence rate by ethnic group was compared using Fishers’ exact test. Age-standardisation was performed using direct standardisation to the WHO standard population 2000–2025; 13 95% confidence intervals for standardised rates were calculated using nonparametric bootstrap estimation with 500 replications. The Stata module RECAP was used to perform a capture-recapture analysis to estimate the number of prevalent cases missing from the data using three case ascertainment. 11
Literature review of international incidence and prevalence rates
We provide a survey of previous epidemiological studies of SMA with which we have compared our data, in the Supplementary Material. We also provide there the detail of how we have approached selection, inclusion and analysis of the data.
Results
Patients identified
Eighty patients with SMA were identified as either being incident in the observation period (19 patients) or living in Aotearoa-New Zealand on the study census day (71 patients). Of the 19 incident patients just ten were also counted in the prevalence cohort, nine having died or moved overseas (see also Supplementary Table 1). There were 58 singletons, ten affected siblings-pairs and two who had an affected sibling who were not included.
These 80 patients were distributed amongst SMA types as follows: SMA0 (1), SMA1 (9), SMA2 (31), SMA3 (37), SMA4 (2). Sex ratio was almost exactly half-and-half male:female Just 3.9% of the sample identified as Māori compared with 16.5% of people in the general population (Table 2). Seventy-nine of the 80 patients had homozygous SMN1 deletion with just one person found to be homozygous for a point mutation. One person's genetic diagnosis was attributed on the basis of their similarly affected sibling's positive diagnosis – a practice not-uncommon in genetic diseases in the past when costs of genetic testing were higher than more recently. There were just 9 cases with a known SMN2 copy number, of which 2 cases had 2 copies (1 with SMA2 and 1 with SMA3) and 7 cases had 3 copies (4 with SMA2, 2 with SMA3 and 1 with SMA4).
SMA subtypes and demographics of whole study sample.
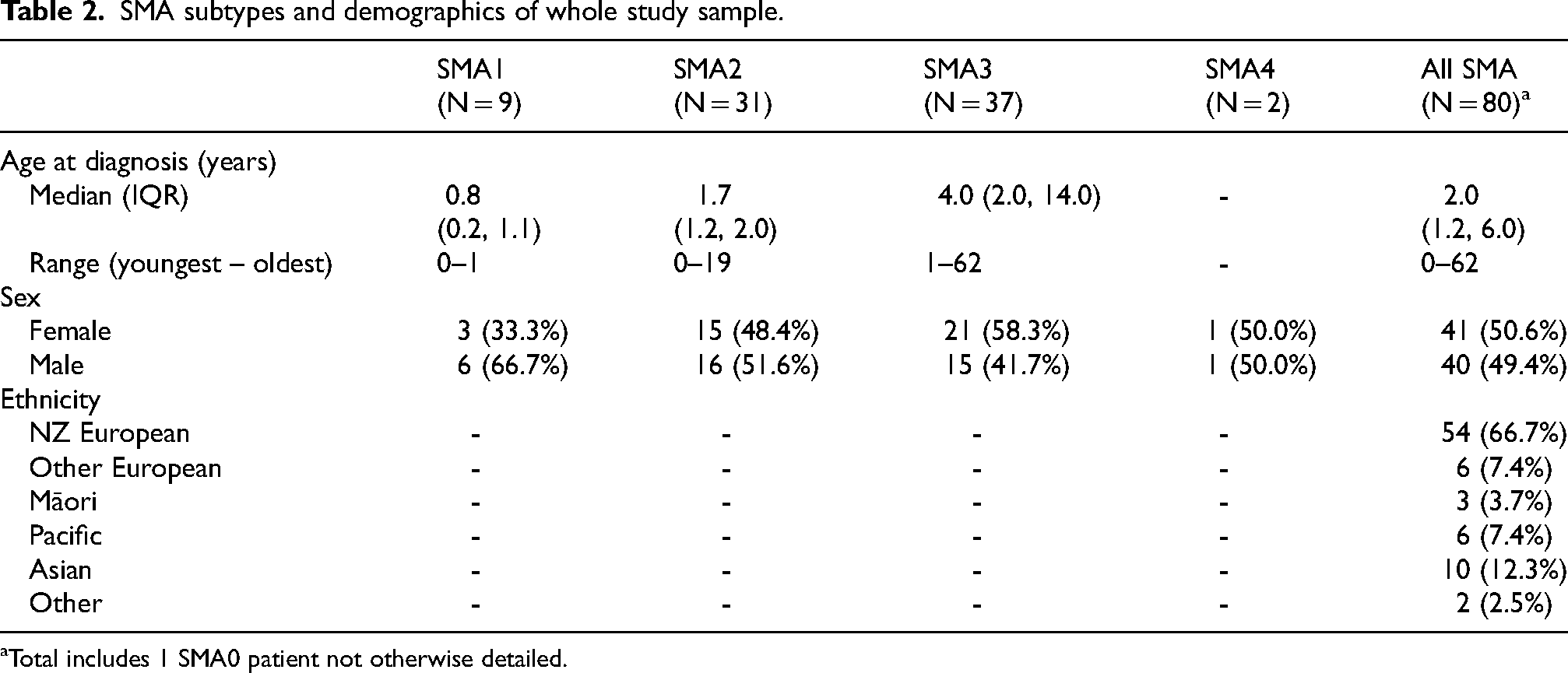
Total includes 1 SMA0 patient not otherwise detailed.
Incidence
The average annual incidence was 8.0 (4.8–12.5, 95% CI) per 100,000 live births (Table 3). The rate was reasonably consistent over the four years. Roughly a third of patients were diagnosed with each of SMA1, 2 and 3 with just one additional patient diagnosed with type 0 and no type 4. Fourteen were NZ European/Other European, 1 Māori, 2 Pacific People, 1 Asian and 1 “Other”. Incidence rates per 100,000 live births (95% CI) by ethnicity were: NZ European/Other European/Other 13.5 (7.6, 22.3); Māori 1.7 (0.0, 9.3); Pacific People 8.3 (1.0, 30.0) and Asian 2.4 (0.0, 13.2) (see Supplementary Table 3).
SMA incidence in NZ, comprising 19 cases diagnosed in NZ between 1st July 2015 and 30th June 2019.
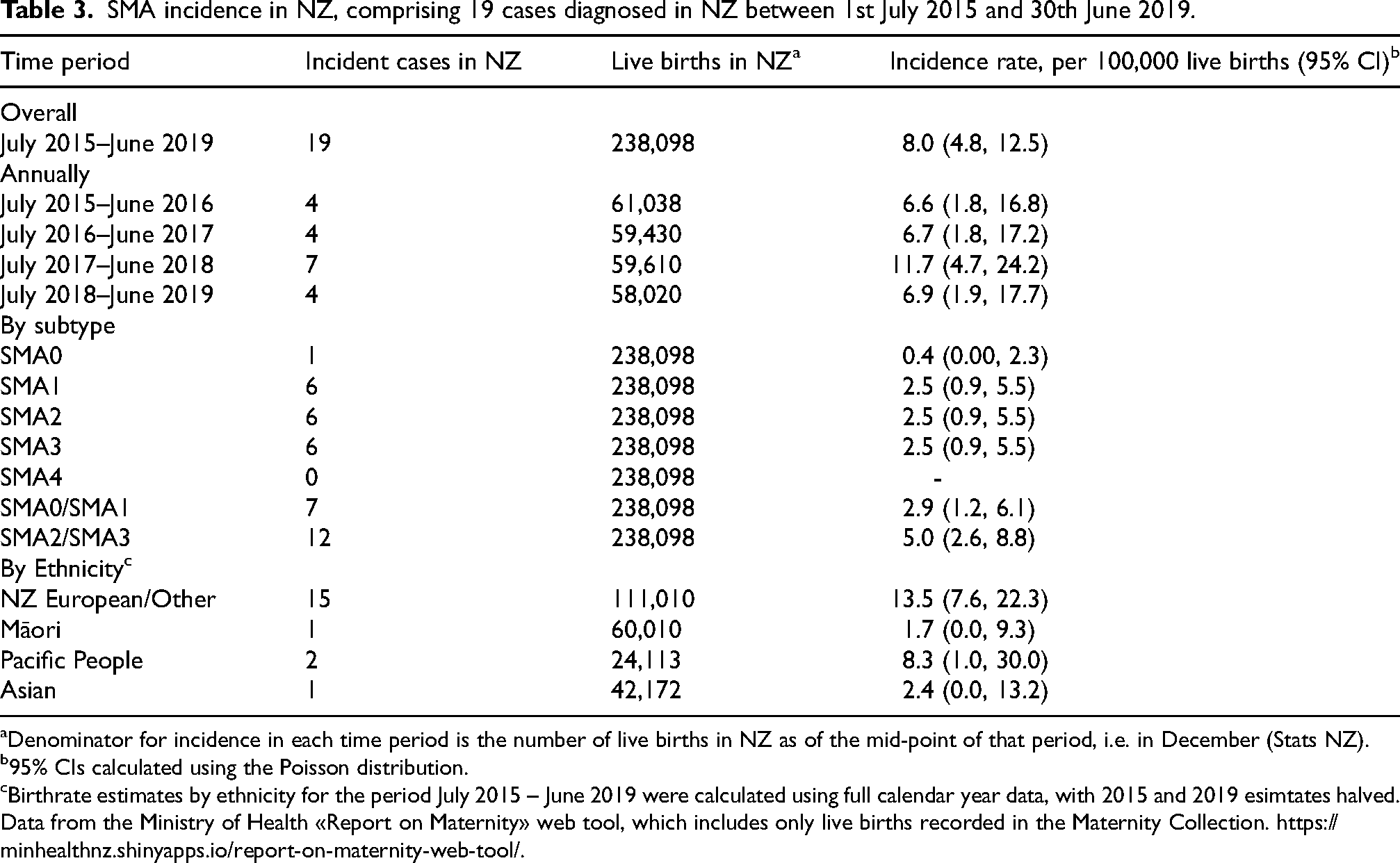
Denominator for incidence in each time period is the number of live births in NZ as of the mid-point of that period, i.e. in December (Stats NZ).
95% CIs calculated using the Poisson distribution.
Birthrate estimates by ethnicity for the period July 2015 – June 2019 were calculated using full calendar year data, with 2015 and 2019 esimtates halved. Data from the Ministry of Health «Report on Maternity» web tool, which includes only live births recorded in the Maternity Collection. https://minhealthnz.shinyapps.io/report-on-maternity-web-tool/.
Prevalence
The overall standardised prevalence rate of SMA in Aotearoa-New Zealand on 1st March 2019 was 1.78 per 100,000 (95% CI: (1.24, 2.33)) (Table 4). Of the 19 cases in the incidence analysis, seven (one with SMA0, six with SMA1) had died before the prevalence date. Five were < 6 months old at time of death, and two between 6 months and 3 years old). A further two patients (one SMA2 one SMA3) had moved overseas (to access disease modifying therapy). The overall median age for all 71 patients was 19.0 years (interquartile range 8.0,28.0). The median age for each of the subtypes were 6.0 years (5.0, 8.0) for SMA1; 14.0 years (6.0,25,0) for SMA2; 21.0 years (15.0, 36.0) for SMA3 and 27.5 years for SMA4.
SMA prevalence for n = 71 cases alive and living in NZ on 1st March 2019.
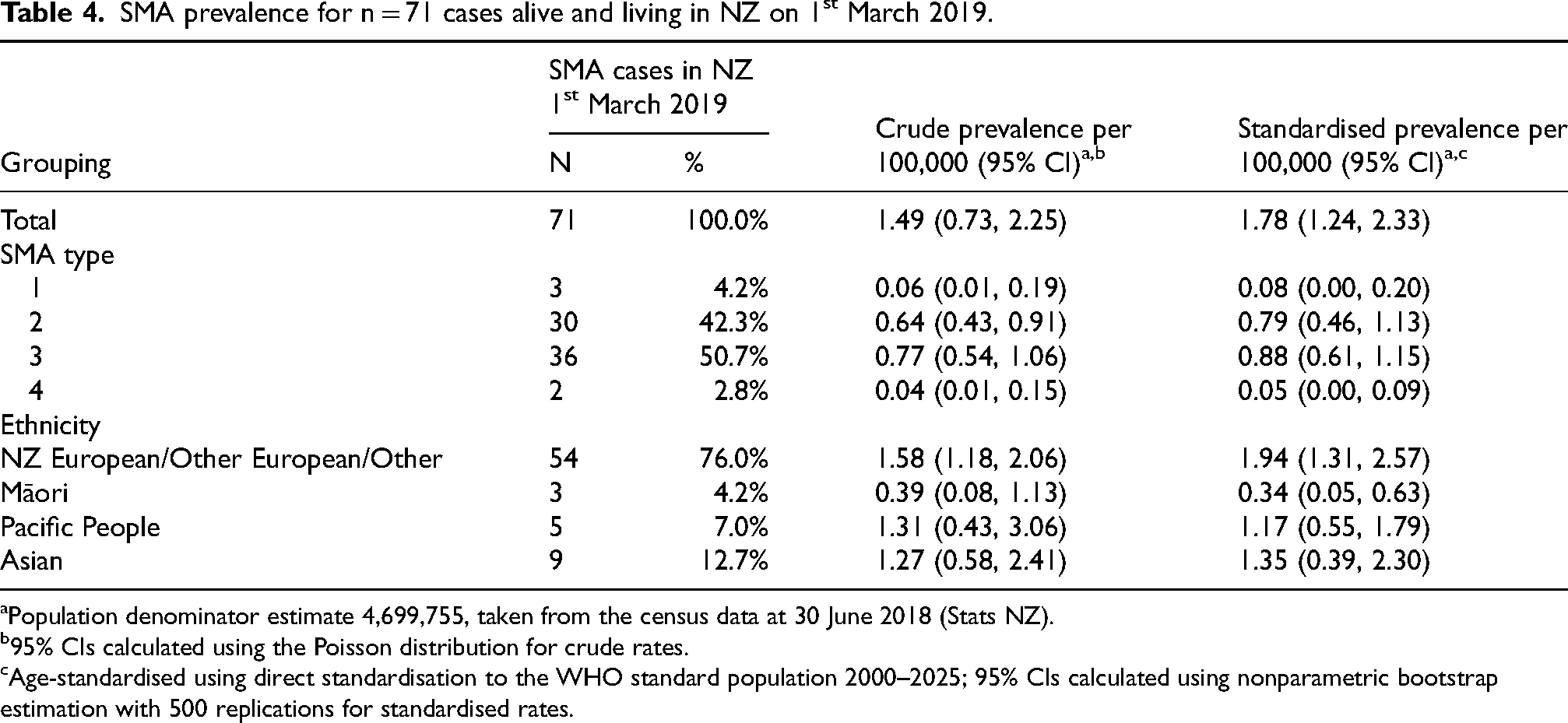
Population denominator estimate 4,699,755, taken from the census data at 30 June 2018 (Stats NZ).
95% CIs calculated using the Poisson distribution for crude rates.
Age-standardised using direct standardisation to the WHO standard population 2000–2025; 95% CIs calculated using nonparametric bootstrap estimation with 500 replications for standardised rates.
As suggested by the incidence results, a substantial and statistically significantly (Fishers’ exact test p = 0.006) lower prevalence rate was also seen amongst Māori – about one-sixth that seen in the population overall (Table 4). Prevalence rates in Pacific people and Asians were slightly lower than that for Europeans/others; however, these differences were not statistically significant (p = 0.83 and p = 0.09, respectively), and estimates for these three ethnic groups were broadly consistent with the average rate for the country (Figure 1).
We explored the possibility that patients in more remote settings might have a lower likelihood of diagnosis. We found no evidence to support this with just one region (Wellington - an urban centre) where the 95% CI did not cross the overall average (see Supplementary Figure 1). The result for Wellington probably just reflects random variation in a small sample.
Capture-recapture analysis
We received data from four different data sources (Supplementary Table 3 and Supplementary Figure 2). The vast majority of cases (97.2%) were identified from doctor records. The Needs Assessments Agencies identified only two cases, which were both also identified in two or three other sources. Cases identified via Pūnaha Io – the NZ NeuroGenetic Registry and Biobank were all also identified through another source. All but three cases had at least two sources of ascertainment. In two of these cases the only source of identification was via the lab, and in the third, the only source was doctor records. A capture-recapture analysis in Stata using the 3 main lists (doctors, lab and NMD Registry – total N = 71) gave an estimated N of 71 (95% CI 71,74). 11
Capture-recapture analysis relies on i) different sources identifying the same person in a way that enables us to know they’re the same person – for our dataset all sources use the same National Health Index number ii) accounting for transient patients might appear in one dataset but not another, as the point prevalence date was the same for all databases this did not occur; iii) Patients have an equal chance of being identified by one of the methods. This very high response rate from New Zealand neurologists, the national coverage of Pūnaha Io and the fact that all patients diagnosed in New Zealand would have had their samples assessed in one of the three laboratories satisfies this criterion. Patients diagnosed outside New Zealand who never consulted a neurologist would not have been picked up this way but these are likely to be very few.
Disability and health resource utilisation
The data from the prevalence sample are presented here. Only three SMA1 patients were alive in our cohort at the point prevalence date with a median age of 6 years (see Table 5). All definitely satisfied the diagnostic criteria for SMA1 in that they had symptom onset before six months of age, and never learned to sit independently. Amongst the SMA2 patients, a range of best motor milestones was seen. All had achieved independent sitting. The median maximum motor milestone achieved was only just greater than that at crawling; the upper quartile range was standing with support. All type 3 and 4 patients had achieved independent walking but there had been substantial decline amongst the SMA3 group with almost half requiring support even to sit by prevalence day. Of the patients with SMA3a (onset before age 3), 68% were permanent wheelchair users and 14% were intermittent wheelchair users. Amongst patients with SMA3b the respective figures were 8% and 17%. Both SMA4 patients were still ambulant (See Supplementary Table 2).
Maximum and prevalence day motor milestones, SMA complications and interventional management in prevalence sample.
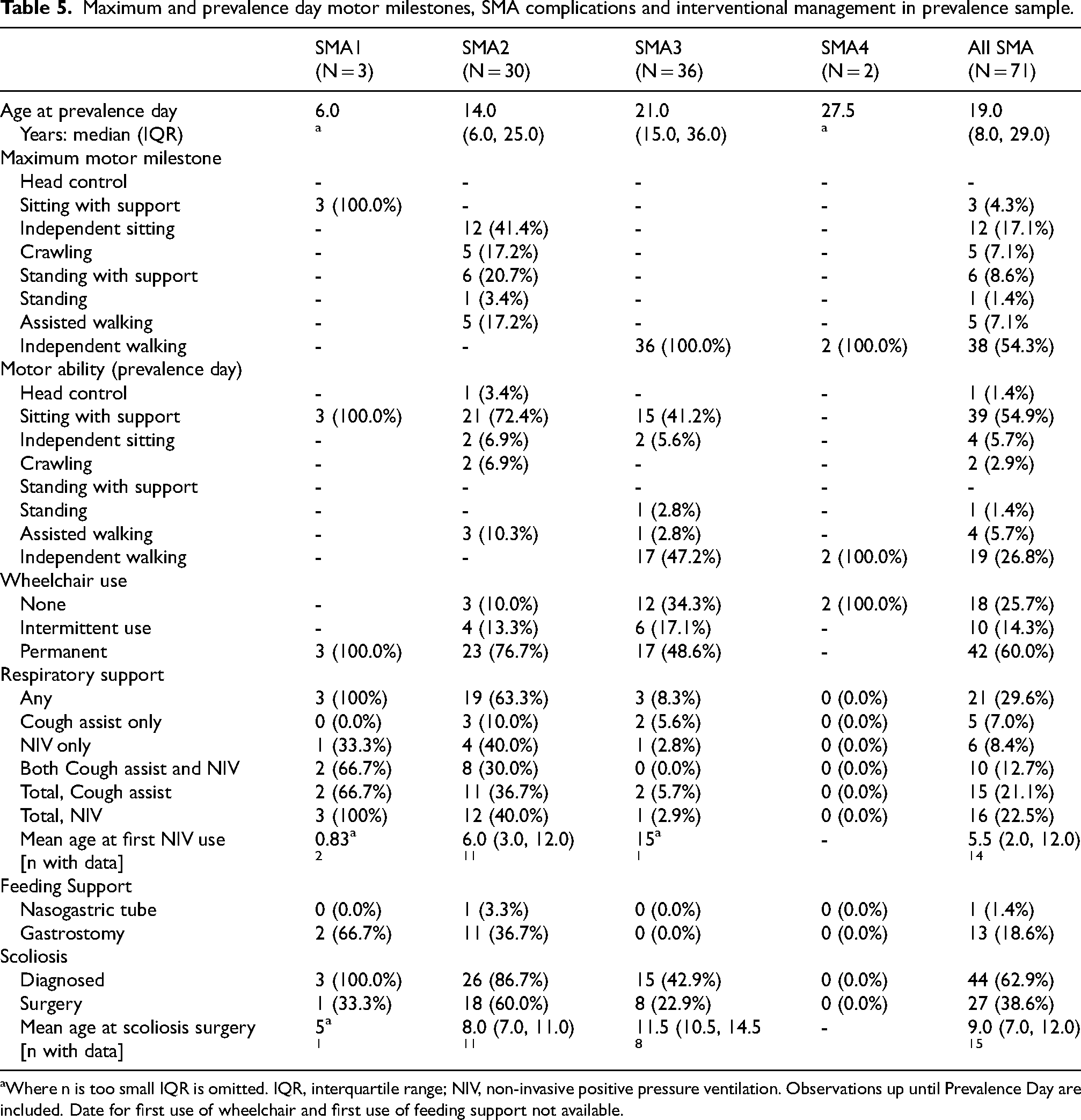
Where n is too small IQR is omitted. IQR, interquartile range; NIV, non-invasive positive pressure ventilation. Observations up until Prevalence Day are included. Date for first use of wheelchair and first use of feeding support not available.
Almost a third of all patients used either cough assist devices, non-invasive ventilation (NIV) or both (Table 5), including all patients with SMA1 and 40% of those with SMA2. Positive pressure ventilation was seldom required in SMA3 (n = 1 patient with SMA3a), and not used at all in SMA4. Tube feeding was required for 40% of patients with SMA2, almost all of whom had a gastrostomy inserted for this purpose. No patients with SMA3 or 4 required tube feeds. 62% of all SMA patients developed a scoliosis, including all patients with SMA1, 87% of patients with SMA2, and 43% of patients with SMA3 (including two SMA3b patients). Over half the patients with scoliosis (61%) required spinal surgery. For those for whom data were available, the median age of first NIV use was 5.5 (2.0, 12.0) years and scoliosis surgery 9.0 (7.0,12.0) years (Table 5).
Utilisation of healthcare services was high across all SMA subtypes. The three SMA1 patients had had a median 1045 out-patient appointments (medical and allied health), 22 inpatient admissions and 6 visits to theatre each (Table 6). SMA2 patients also had very high healthcare demands with the 31 patients undergoing a median 89 out-patient visits, 7 inpatient visits, and 2 theatre visits. The 37 SMA3 patients still required significant resources with a median 33 outpatient visits each as well as one in-patient admission and one theatre visit.
Lifetime per-patient reported hospital visits for prevalence sample.
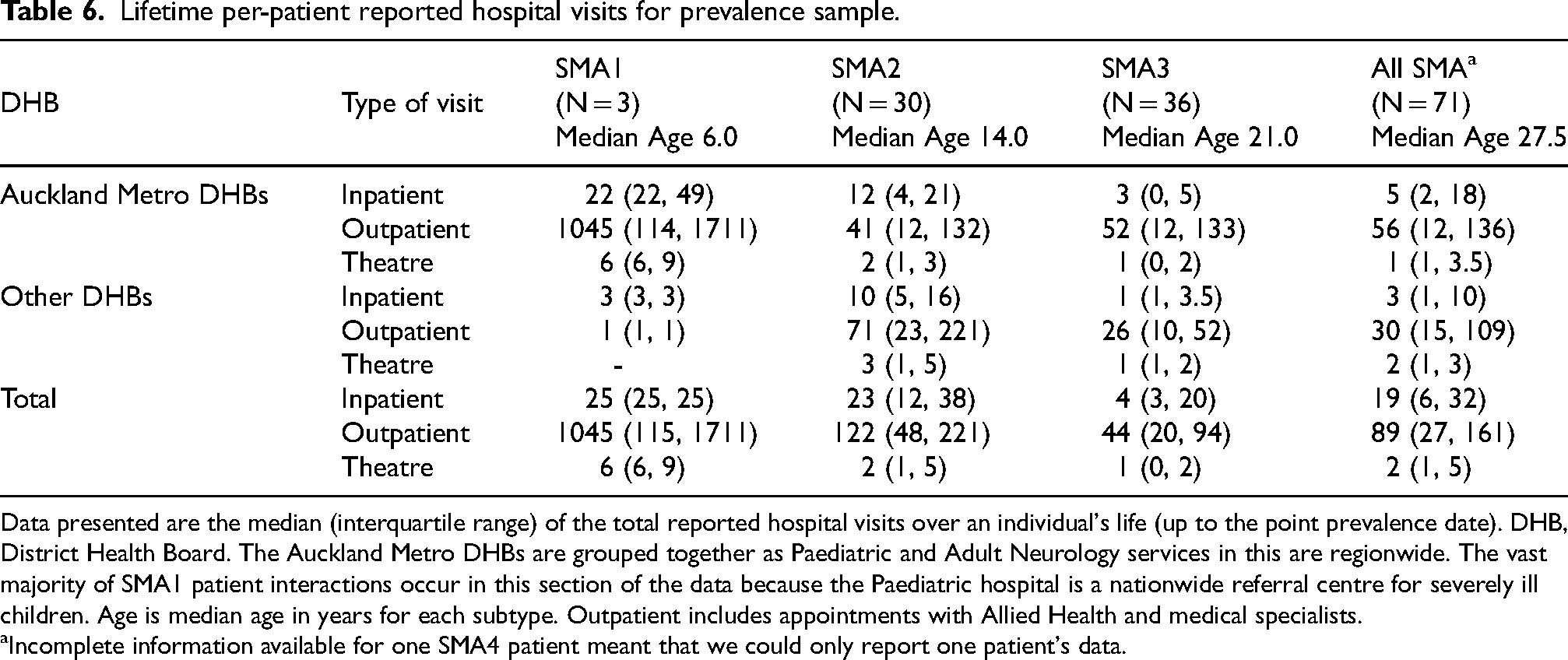
Data presented are the median (interquartile range) of the total reported hospital visits over an individual's life (up to the point prevalence date). DHB, District Health Board. The Auckland Metro DHBs are grouped together as Paediatric and Adult Neurology services in this are regionwide. The vast majority of SMA1 patient interactions occur in this section of the data because the Paediatric hospital is a nationwide referral centre for severely ill children. Age is median age in years for each subtype. Outpatient includes appointments with Allied Health and medical specialists.
Incomplete information available for one SMA4 patient meant that we could only report one patient's data.
Discussion
The advent of three effective disease modifying therapies for SMA has highlighted the need to understand the epidemiology of SMA for and its disability impact to inform healthcare planning. We present here the results of a thorough retrospective, four-year incidence, and point prevalence study of SMA in the whole population of Aotearoa-New Zealand. The incidence of SMA was 8.0 cases per 100,000 live births (95% CI: (4.8, 12.5)), which places Aotearoa-New Zealand in the middle of international estimates of prevalence. SMA subtype incident rates were also in keeping with the literature. The age standardised prevalence on 1st March 2019 was 1.78 per 100,000 (95% CI: (1.24, 2.33)) – near the high end of the range of previous studies. However, other studies have not provided age standardised results which makes it difficult to make full comparisons. Age-standardised prevalence figures for SMA subtypes were also generally consistent with previously observed rates though our study found a higher SMA2 prevalence rate than the two previous studies. We are the first study to give a prevalence estimate for SMA4.
Capture-recapture analysis suggests that all patients with a diagnosis of SMA in Aotearoa-New Zealand were found. While it is possible that not all patients with SMA are diagnosed, infants with SMA1 are likely to present to paediatricians and neurologists because of the severity of their disease and genetic testing is widely available and publicly funded so it is likely that most patients with SMA1 have been found in our study. However, because genetic testing for SMA has only been readily available in Aotearoa-New Zealand since 2007 it is possible that older patients with SMA3 and SMA4 have not been diagnosed. This would be consistent with the upper interquartile range for SMA3 being just 36 years. With the introduction of disease modifying therapies more patients with adult SMA may present for diagnosis.
Almost all incidence and prevalence rates (totals and SMA subtypes) from our study lie within the range of those previously observed (Table 7). The only exception was our estimate for the standardised prevalence of SMA2, (0.79/100,000, 95% CI 0.46–1.13) which was higher than the two previous estimates (0.13 and 0.53). It is also interesting to note the total SMA rates are within the range of those reported in large newborn screening programmes such as in a Russian study 12.8/100,000 (26 out of 202 908 live births 16 and national statistics in the USA of 6.8/100,000 (425 out of 6 244 825) live births. 17
Present study incidence and prevalence compared with literature.

SI, Standardised Incidence; SPP, Standardised point prevalence; CI, confidence interval (see Supplementary Tables 5 and 6) – note the figures above are based on the following papers.14,18–46
Of particular note, our “All SMA” prevalence estimate of 1.78/100,000 was close to the most similarly conducted prevalence study by Norwood et al. 14 which showed a prevalence of 1.9/100,000. Generally, our SMA1 incidence and prevalence were at the lower level of that reported whereas our combined figures for SMA2 and 3 almost exactly mirror the literature. We are the first to report a prevalence for SMA4 and the only study to report population standardised data.
This study was performed at a time when New Zealand patients did not have access to disease modifying therapies. It was notable that many of the patients identified in the incidence part of the study were no longer living in Aotearoa-New Zealand on prevalence day. Seven patients with SMA0 or 1 died without access to therapy, and two patients had moved overseas in the hope of receiving disease modifying therapy. Both of these outcomes are devastating and potentially preventable with funded access to powerful RNA based therapies such as nusinersen and risdiplam and also gene therapy with onasemnogene abeparvovec. Furthermore, it gives some context to the surprising survival of three infants with SMA1 beyond the natural history expected for infants with SMA1 – these are the minority - prevalence studies naturally select patients with longer survival.
While no study has found a population free of SMA, disease prevalence and carrier frequencies have been shown to differ across ethnic groups.15,18 The substantially lower observed prevalence amongst New Zealand Māori where an overall standardised prevalence of 0.34 cases per 100,000 people was seen may be another example of this. Alternatively, it could represent under-reporting due to the known barriers to healthcare for Māori.47,48 Our incidence figures were consistent, and the finding of higher rates amongst Pacific People (almost three times higher) and rates in rural areas similar to urban (Supplementary Figure 1 footnotes) – both populations known also to experience barriers to healthcare in Aotearoa-New Zealand49,50 suggest that this is likely to be a true finding. This is important within Aotearoa-New Zealand from points of view of equity for our indigenous people and for future projections of healthcare needs as the proportion of Māori in the population increase (Stats NZ).
Another important finding of our study is that one in six families had more than one affected sibling. While government-funded genetic counselling is available to all families affected by a diagnosis of SMA and two cycles of IVF/preimplantation genetic diagnosis have been funded since 2005, in the vast majority of families, the couple was already pregnant or the second child born by the time their older child was diagnosed. The potential to avoid a second affected pregnancy highlights the benefit of new-born or preconception screening. 51
This study quantifies the very significant direct health impacts of untreated SMA on the medical system in New Zealand. Patients with SMA1 had especially high numbers of outpatient appointments. Healthcare encounter rates were progressively lower in SMA2 and SMA3 but given the overall numbers of prevalent patients, still represent a significant health resource and individual burden. The need for support with ambulation/mobility, ventilation and scoliosis management was very high across all patients. Three quarters required assistance with ambulation most commonly requiring a wheelchair. Almost 30% required ventilatory support and almost 20% ongoing tube feeding. Scoliosis affected almost two thirds and close to 40% underwent scoliosis surgery. Standards of Care guidelines are vital in ensuring patients have active surveillance and management of these complications. This becomes increasingly important, alongside disease modifying therapy, to maximise the benefits of treatment as we seek to best understand the new phenotype of treated SMA. Disease modifying therapies offer the best outcomes with early, ideally presymptomatic access to treatment in order to prevent the irreversible loss of motor neurones.52,53 Increasing health economic analyses support newborn screening and pre-symptomatic treatment as the best way to reduce morbidity and health care burden.15,54,55
The main potential weakness of the study is its retrospective nature. Determination of current clinical status and motor milestone achievement for each patient was limited by the frequency at which medical review and milestone assessments were performed. Although this is the one of the largest studies to date where clinical diagnosis (not just genetic status) was individually verified, the total numbers are relatively small. However, these issues are offset by a number of features. In particular, we calculated the prevalence on a whole population basis and aimed to identify not just children but adults with SMA. The fact that our study found patients through multiple sources allowed for a capture-recapture analysis which gave reassuring results as to the comprehensiveness of the study. It is the first study to calculate standardised prevalence rates, which will allow for more accurate international comparisons with future studies.
In conclusion, overall the prevalence of SMA in New Zealand is similar to international figures. The substantially lower prevalence and incidence seen in Māori in this study warrants further investigation and will be one of the outcomes that may be assessed when newborn screening is introduced. The study also underscores the high rates of morbidity and heavy use of health resources associated with untreated SMA - highest amongst patients with SMA types 1 and 2, but still substantial amongst types 3 and 4. Going forward, the quantification of morbidity will provide a baseline for evaluating of the impact of disease modifying therapy when used to treat symptomatic or pre-symptomatic infants.
Supplemental Material
sj-pdf-1-jnd-10.1177_22143602251319165 - Supplemental material for Epidemiology of spinal muscular atrophy in Aotearoa-New Zealand
Supplemental material, sj-pdf-1-jnd-10.1177_22143602251319165 for Epidemiology of spinal muscular atrophy in Aotearoa-New Zealand by Richard H Roxburgh, Alana Cavadino, Miriam Rodrigues, Sharron Meadows, Juliette Meyer and Gina O'Grady in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
The original literature search and strategy was devised by Prof Alice Theadom and we learned much from her approach to neuroepidemiology.
Funding
The study was funded by Biogen (Australia New Zealand) as an Investigator initiated project.
Competing interests
RR, MR and GO’G have served on an NZ Advisory board for Risdiplam (Roche).
MR and GO'G have served on an NZ Advisory board for nusinersen (Biogen).
Data availability statement
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.