
Abstract
Oculopharyngodistal myopathy (OPDM) is a rare muscular disorder characterized by ocular symptoms, pharyngeal symptoms, facial weakness, and distal predominant limb muscle weakness. The cause of the disease was unknown for a long time. Recently, however, it has been reported that expansions of CGG or CCG repeats in LRP12, LOC642361/NUTM2B-AS1, GIPC1, NOTCH2NLC, RILPL1, and ABCD3 are the causes of the disease. Cases sometimes present with neurological symptoms, and the clinical spectrum of diseases caused by expansions of CGG or CCG repeats has been proposed to be called FNOP-spectrum disorder after the names of fragile X-associated tremor/ataxia syndrome, neuronal intranuclear inclusion disease, oculopharyngeal myopathy with leukoencephalopathy, and OPDM. In this article, the recent progress in the field of OPDM is reviewed, and remaining issues in OPDM are discussed.
Keywords
Oculopharyngodistal myopathy (OPDM) is a rare muscular disorder characterized by ocular symptoms (ptosis and limited eye movement), pharyngeal symptoms (dysarthria characterized by nasal voice and dysphagia), and distal predominant limb muscle weakness. OPDM patients also have facial weakness. The disorder usually shows an autosomal dominant mode of inheritance, but sporadic cases are frequently observed. The cause of the disease was unknown for a long time, and the diagnosis was often limited to clinical diagnosis. Recently, however, it has been reported that expansions of CGG or CCG repeats in several genes are the causes of the disease, and the pathogenesis of the disease is rapidly becoming clear. In this article, the recent progress in the field of OPDM is reviewed.
History of OPDM
OPDM was first reported in 1977 by Satoyoshi and Kinoshita, 1 in which the clinicopathological features of four families were described. The following year, Jasper et al. reported a similar disease from Europe, 2 and the family with this disease was later described in detail again by van der Sluijs et al. 3 As shown in Figure 1, as of 2019, most cases of this disease have been reported from Japan, China, Turkey, and Southeast Asia1,4–9; reports from other regions were extremely rare.2,3,10–12
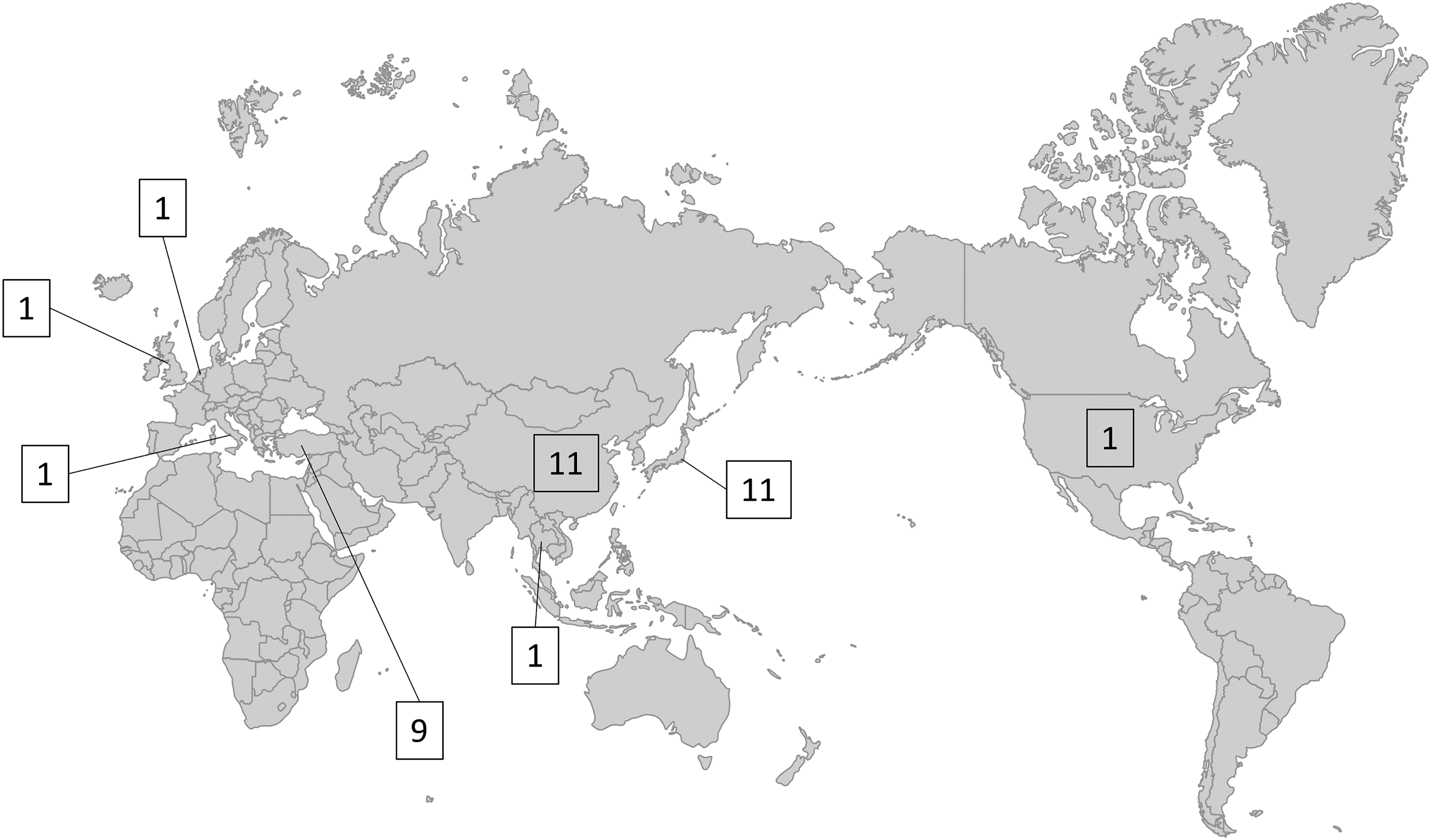
Number of reported OPDM families as of 2019. This figure shows the number of OPDM families as of 2019. Only 14 independent case reports in 42 years have been published until 2019. There are relatively many reports from Japan, China, and Turkey.
In 1998, PABPN1 was reported to be the causative gene of oculopharyngeal muscular dystrophy (OPMD). 13 Most cases of OPMD are caused by an abnormal expansion of GCG/GCA repeats in the (GCG)6(GCA)3(GCG)1 sequence, which encodes 10 polyalanines from the second to 11th codon, in exon 1 of PABPN1. In patients with OPMD, the polyalanine tract is usually expanded to more than 12 repeats or has 11 repeats in a homozygous state. Although rare, a case of 13 polyalanine repeats due to the point mutation p.Gly12Ala including the 13th and 14th alanine codons has been reported. 14 Recently, it has been reported that the p.Gly12Trp mutation also causes OPMD. 15
After the discovery of PABPN1, clinical differences between OPMD and OPDM have been recognized. Although both have ocular, pharyngeal, and limb muscle weakness as the main symptoms, OPDM is characterized by distal predominant limb muscle weakness, whereas OPMD is characterized by proximal predominant limb muscle weakness. In addition, facial muscle involvement tends to be more prominent in OPDM. The muscle pathology is characterized by the presence of myogenic changes with rimmed vacuoles in both diseases, but it was noted that tubulofilamentous inclusions of 8.5 nm diameter are characteristic in OPMD. 16 Thus, OPDM had only been diagnosed on the basis of the distributions of affected muscles, muscle biopsy findings such as rimmed vacuoles without OPMD-specific 8.5-nm-diameter tubulofilamentous inclusions, and the exclusion of OPMD by genetic analysis.4,5
Brief history of the identification of causative genes of OPDM
The identification of the causative gene for OPDM has long been difficult because the disease is rare, it is almost impossible to analyze a large family, and there are many sporadic cases, which means that the mode of inheritance is unclear and/or that the penetrance is not high. The situation changed after the discovery of a family with oculopharyngeal myopathy with leukoencephalopathy (OPML), which presented with oculopharyngeal myopathy but was also complicated by leukoencephalopathy resembling symptoms found in neuronal intranuclear inclusion disease (NIID) and autonomic dysfunctions. 17
NIID is an autosomal dominant or sporadic neurodegenerative disease.18,19 There are two main clinical types: cases predominantly with peripheral neuropathy and cases predominantly with cognitive dysfunction and fluctuating consciousness (encephalitis-like episodes) as the main symptoms. 18 The peripheral neuropathy-predominant form tends to have a younger age of onset. Pathologically, the disease is characterized by the presence of ubiquitin- and p62-positive inclusions in the nuclei of neurons and glial cells. These inclusions are also present in the skin and various organs of the body, allowing antemortem diagnosis. 20 Leukoencephalopathy is usually present in the dementia-predominant type, and hyperintensities in corticomedullary junctions are the hallmark of this disease on diffusion-weighted images. Other neuroimaging features include hyperintensities in the middle cerebellar peduncles and paravermal regions in T2/FLAIR images. During and after an encephalitis-like attack, the diffusion-weighted imaging (DWI) hyperintensity is enlarged and is swollen and later shows atrophic changes. During this time, the cerebral blood flow is considered to be locally enhanced and later reduced.
On the basis of the finding that the clinicopathological presentations of NIID are similar to those of fragile X-associated tremor/ataxia syndrome (FXTAS), which is caused by 55–200 CGG repeats (premutation) in FMR1, we hypothesized that NIID is also caused by an expansion of CGG repeats in another gene. We attempted to directly extract expanded CGG repeats from whole-genome sequencing data and found expansions of CGG repeats in NOTCH2NLC (formerly annotated as NBPF19) in patients with NIID. The expanded CGG repeats are found in not only familial cases but also sporadic cases.
Subsequently, we hypothesized that OPML is also caused by expansions of CGG repeats because the neuroradiological findings of OPML were similar to those of NIID. By extracting expanded CGG repeats directly from the short-read whole-genome sequencing data, we found that OPML is caused by an abnormal expansion of CGG repeats at the site where two non-coding genes, LOC642361 and NUTM2B-AS1, are expressed in both directions. The form of OPML in this family was designated as OPML type 1.
Finally, we assumed that OPDM was also caused by expansions of CGG repeats in other genes, because oculopharyngeal myopathy was common in OPML and OPDM. Whole-genome sequencing data from OPDM families revealed an expansion of CGG repeats in the 5’ untranslated region of LRP12. Additional analysis revealed the expanded repeats in 22 patients with familial and sporadic OPDM, whereas only 0.2% of control subjects had the expansions. Thus, we concluded that the expanded CGG repeats in LRP12 were pathogenic for OPDM. 17 This form of the disease is referred to as OPDM type 1. In some cases, this repeat expansion was not detected, suggesting further genetic heterogeneity.
Subsequently, linkage analysis, repeat-primed PCR analysis, and long-read sequencing of large OPDM families in China identified expansions of CGG repeats in the 5’ untranslated region (UTR) of GIPC1.21,22 Such expanded repeats were also found in Japan. 1 This form of the disease is referred to as OPDM type 2.
Reports from Japan and China revealed that some patients with OPDM symptoms had expansions of CGG repeats in NOTCH2NLC, the causative gene of NIID, and this form is termed OPDM type 3.23,24 Some patients also have hyperintensities in the white matter on brain T2-weighted images and hyperintensities on diffusion-weighted images, as seen in patients with NIID. That is, the combination of leukoencephalopathy and oculopharyngeal myopathy is not limited to patients with OPML1, but also in those who have expansions of CGG repeats in NOTCH2NLC.
Two studies from China showed that expansions of CCG repeats (in the direction of RILPL1 transcription) immediately upstream of RILPL1 also cause OPDM25,26; analysis of RNA-seq data has shown that this site, previously considered to be upstream of the transcription start site, is in fact transcribed and is concluded to be located in the 5’ untranslated region of RILPL1. Antisense transcription was also found in RNA sequencing. 26 This form of the disease is referred to as OPDM type 4. Another large pedigree with OPDM type 4 was also reported. 27
Until recently, only one family with expansions of CGG or CCG repeats in LOC642361 or NUTM2B-AS1, respectively, was known. 17 Two groups, however, recently reported cases of new families. Two cases of patients were reported from a Chinese group. 28 Very interestingly, these two patients did not have leukoencephalopathy, but showed an OPDM phenotype. Another group reported the case of 12 patients from three families with expansions of CGG or CCG repeats in LOC642361 or NUTM2B-AS1, respectively. 29 Only one patient showed hyperintensities in the middle cerebellar peduncles, suggesting neurological involvement of the diseases. Another paper showed that expansions of CGG or CCG repeats in LOC642361 or NUTM2B-AS1, respectively, was found in four patients from three Thai families, 55 including that reported before. 6 In two patients, brain MRI showed T2 hyperintensity along the corticospinal tract; however, extensive white matter lesions, as observed in the first OPML patient, were not present. This suggests that patients with repeat expansions in LOC642361/NUTM2B-AS1 are more likely to exhibit a clinical phenotype of OPDM rather than OPML.
In a European population, expansions of CCG repeats in the 5’ UTR of ABCD3 have recently been identified by Cortese et al. 30 The number of CCG repeats ranged from 118 to 694 repeats. They found that ABCD3 transcripts were upregulated, suggesting the gain-of-toxic effect of the expanded repeats.
Molecular epidemiology of OPDM
Cases of OPDM continue to be reported mostly from East Asia. In Japan, suspected cases of OPDM or OPMD between 1978 and 2021 included 134 OPMD, 70 OPDM1, 12 OPDM2, and 7 OPDM3 cases; there were no OPDM4 cases. 31
A paper on 51 families from China reported OPDM1 in 3.9%, OPDM2 in 37.3%, OPDM3 in 13.7%, OPDM4 in 21.6%, and cases of unidentified cause in 23.5% of these families. This distribution is characterized by fewer OPDM1 cases, and more OPDM2 and OPDM4 cases than in Japan. 25
In families of European descent, expansions of CCG repeats in ABCD3 have just been reported. 30 No other studies have been reported regarding OPDM1, OPDM2, OPDM3, OPDM4, or OPML1 in the population.
Clinical characteristics of OPDM
The clinical characteristics of OPDM are ocular, facial, pharyngeal, and distal predominant limb weakness. OPDM typically presents in middle age and follows an extremely slow progression. Diagnosis is relatively straightforward when all clinical features are present; however, in patients who exhibit only partial symptoms over a prolonged period, diagnosis can be challenging and may take considerable time. Although cardiac complications and respiratory failure are uncommon, it is essential to monitor elderly patients for aspiration pneumonia due to dysphagia as the disease progresses.
Since the causative genes were identified, more detailed clinical analyses have become possible. Here, the clinical characteristics of each disease type and new findings on differentiating features between OPDM and OPMD are discussed.
Muscle biopsy findings
Typical histopathological features of OPDM include myogenic changes with rimmed vacuoles. This finding is common in both in OPMD and OPDM, although it has long been said that the 8.5 nm nuclear inclusion body is characteristic of OPMD, whereas it is absent in OPDM. 4
In an extensive clinicopathologic analysis of 65 patients with OPDM, electron microscopy findings of OPDM1 were reported by Kumutpongpanich et al.. 32 They found nuclear abnormalities including hyperchromicity and an irregular, fragmented appearance of nuclei in all patients examined, although these findings were observed only in less than 1% of the nuclei. One patient showed tubulofilamentous intranuclear inclusions, whose diameter was 17.3 ± 1.4 nm, and one patient showed cytoplasmic filaments of approximately 10 nm diameter. In terms of inclusions, 16–18-nm-diameter cytoplasmic tubulofilamentous inclusions 9 in addition to intranuclear inclusions 21 were found in patients with OPDM2. In a patient with OPDM3, 12.6 ± 1.6-nm-diameter intranuclear inclusions were found. 33 In OPDM4, intranuclear inclusion bodies were found in the muscle25,26 and fibroblasts. 26
Ogasawara et al. examined p62 expression in muscle specimen by immunohistochemistry in 19 patients with OPDM1, 6 with OPDM2, 7 with OPDM3, 15 with OPMD, and 37 with other rimmed vacuole myopathies. 33 They found that myonuclei with p62-positive intranuclear inclusions were significantly more frequent in OPMD than in OPDM and other rimmed vacuolar myopathies. On the other hand, intranuclear inclusions in non-muscle cells such as blood vessels, peripheral nerve bundles, and muscle spindles were present in OPDM, but absent in OPMD. The findings suggest a possibility of differentiating OPDM from OPMD.
Muscle imaging study
Eura et al. analyzed clinical characteristics and conducted muscle imaging of 43 patients with OPDM1, 6 with OPDM2, 5 with OPDM3, and 57 with OPMD. 34 The age at onset of OPMD was significantly older than that of OPDM. Neuropsychiatric symptoms were found in 60% of patients with OPDM3 as an initial symptom, whereas other patients showed ptosis, dysphagia, and muscle weakness as initial symptoms. Muscle weakness was distal predominant in 77%, 50%, and 60% of patients with OPDM1, OPDM2, and OPDM3, respectively, whereas it was proximal predominant in 80% of patients with OPMD. In addition to the finding that cases in which distal and proximal muscles are equally affected were observed in both OPDM and OPMD, there were rare cases of proximal muscle weakness in OPDM (0% in OPDM1, 17% in OPDM2, and 0% in OPDM3) and distal muscle weakness in OPMD (3.6%). Muscle imaging revealed that calf muscles were more extensively replaced by fat than thigh muscles in OPDM. The tibialis anterior, extensor digitorum longus, medial head of gastrocnemius, and soleus are particularly affected in OPDM. Among the subtypes of OPDM, OPDM3 showed more severely affected muscles. When comparing OPDM and OPMD, the tibialis anterior and extensor digitorum longus were found to be more affected in OPDM, whereas the adductor magnus was more affected in OPMD.
Intranuclear inclusions in skin biopsy
In NIID caused by expansions of CGG repeats in NOTCH2NLC, it was known even before the genetic mutation was identified that inclusions occur not only in the central and peripheral nervous systems but also in other organs, including the skin. 20 Therefore, a skin biopsy is the first step in diagnosing NIID. To determine whether the skin also contains inclusions in OPDM, Ogasawara et al. examined intranuclear inclusions in skin biopsies in three patients with OPDM1, six with OPDM2, two with OPDM3, and seven patients with other myopathies. 35 They identified p62-positive intranuclear inclusions not only in OPDM3, the disease caused by expanded CGG repeats in NOTCH2NLC, the same gene as in NIID, but also in OPDM1 and OPDM2. However, p62-positive intranuclear inclusions were not always present in all three cell types, namely, sweat gland cells, adipocytes, and fibroblasts, in OPDM2. Two of the three OPDM1 patients did not show intranuclear inclusions. No intranuclear inclusions were found in patients with OPMD, inclusion body myositis, and GNE (bifunctional UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase) myopathy. Thus, p62-positive intranuclear inclusions in the skin are neither specific nor diagnostic for NIID. The frequencies of intranuclear inclusions in OPDM1 and OPDM2, however, are likely to be lower than those in NIID and OPDM3. In a patient with OPDM4, a skin biopsy revealed intranuclear inclusions. 26
Remaining issues in OPDM
Some remaining issues in OPDM are discussed in this section.
Repeat motif–phenotype correlation
As suggested previously,17,36 expansions of CGG or CCG repeats in different genes have been identified as the cause of OPDM. Thus, it seems that the same repeat motif can cause similar diseases. Similar observations have been found in coding regions (expansions of CAG repeats in spinocerebellar ataxias) and noncoding regions (expansions of TTTCA repeats in benign adult familial myoclonus epilepsy).37,38 When a repeat motif–phenotype correlation is found, it is more likely that the expanded repeats themselves are strongly associated with the disease rather than the loss of a certain shared function in the causative genes by expanded repeats. Because there are cases of familial OPDM with unidentified causes, additional expansions of CGG or CCG repeats may be identified in the future.
OPML, OPDM, or FNOP-spectrum disorder?
The index patient in the original OPML family showed oculopharyngeal myopathy and leukoencephalopathy. 17 However, recent studies have also shown several LOC642361/NUTM2B-AS1-related families that exhibit the OPDM phenotype (which may also be referred to as OPDM type 5).28,29 Nowadays, it is better to understand that expansions of CGG or CCG repeats cause a spectrum of diseases, with some patients presenting with NIID-like neurological symptoms, some with OPDM-like muscle symptoms, and sometimes others with a combination of both. In other words, a strict clinical classification of NIID, OPML, and OPDM does not seem to make much sense. With this in mind, we have proposed the clinical term FNOP (FXTAS, NIID, and OPDM)-spectrum disorder. 39 Although many patients appear to present with only neurological or myopathic manifestations, it seems that comprehensive clinical examinations were not performed on such patients. All patients with expansions of CGG and CCG repeats should be evaluated for symptoms ranging from neurological manifestations such as leukoencephalopathy and peripheral neuropathy to oculopharyngeal myopathy.
What causes reduced penetrance?
Very interestingly, expansions of CGG or CCG repeats can be found in not only familial but also sporadic OPDM, indicating reduced penetrance in these diseases. Several considerations have been made as to why the penetrance is low. Intergenerational repeat instability that causes short repeats could explain the low penetrance. 32 It is possible that some cases are very mild and the symptoms are unnoticeable. We had a case in which the repeat length was small and the patient was unaware of symptoms until her 80s, but on examination, she was considered to have OPDM. 40 This situation may be similar to that of myotonic dystrophy.
However, very long expansions of CGG or CCG repeats were more often observed in asymptomatic carriers.21,24,26,32 Some of them showed hypermethylation of the repeats, presumably leading to the silencing of the genes.24,26,41 This situation is similar to that of FMR1. In FMR1, methylation is known to occur when CGG repeats exceed 200 repeat units, resulting in the silencing of the gene. Because FMR1 is located in the X chromosome and the FMRP protein produced by FMR1 is important for brain development, it is considered that very long hemizygous CGG repeats in FMR1 cause fragile X syndrome, not FXTAS. However, gene silencing may not present much of a phenotype if only one allele is silenced in an autosomal chromosome. Further investigation into asymptomatic carriers is necessary to understand the cause of the low penetrance.
Neurogenic or neuropathic involvements in OPDM?
Although rare, some OPDM patients were suspected of having neurogenic changes. 2 After gene identification, some patients with OPDM2 and OPDM3 were found to have neurogenic changes by needle electromyography.24,25 Considering that neuropathy is a relatively frequent finding in NOTCH2NLC-related NIID, 18 it seems reasonable to assume that neurogenic changes in NOTCH2NLC-related OPDM3.
We have also encountered an OPDM1 case with muscle findings that appeared to indicate grouped atrophy or fiber-type grouping. 40 However, clustering of atrophic fibers was observed only in limited fascicles and abundant hypertrophic muscle fibers were observed, which we considered was different from typical grouped atrophy found in muscles of neuropathic etiology. A case of OPDM1 has been associated with peripheral neuropathy, 42 and more recently, CGG repeats in LRP12 are frequently found in patients with a diagnosis of peripheral neuropathy. 43 Most patients with a diagnosis of peripheral neuropathy had motor predominance and lacked sensory deficits. Expansions of CGG repeats in LRP12 have also been found in patients with a clinical diagnosis of ALS. 44 In the paper, signs of upper motor neuron signs appear to be absent or only minimally present in some patients. These studies, however, suggested that electrophysiological and myopathological analysis sometimes reveal a neurogenic and neuropathic pattern in patients with expanded CGG repeats in LRP12. To date, no autopsy findings, especially of the motor nerves or spinal cord anterior horn cells, have been reported for patients with these features. Future studies are needed to clarify this issue.
CGG or CCG repeats, which is important?
The original family with OPML shows expanded repeats in the location where the two genes LOC642364 (CGG direction) and NUTM2B-AS1 (CCG direction) are bidirectionally transcribed. 17 Bilateral transcription in normal muscles was found in stranded RNA-seq. In the same analysis, no antisense transcription was identified in NOTCH2NLC, and trivial antisense transcription was identified in LRP12. Now we know that expanded CGG repeats occur in LRP12 (OPDM1), GIPC1 (OPDM2), and NOTCH2NLC (OPDM3), whereas expanded CCG repeats occur in RILPL1 (OPDM4) and ABCD3 (Table 1). These findings suggest that both expansions of CGG and CCG repeats should be considered as causes of OPDM.
Summary of FNOP-spectrum disorders.
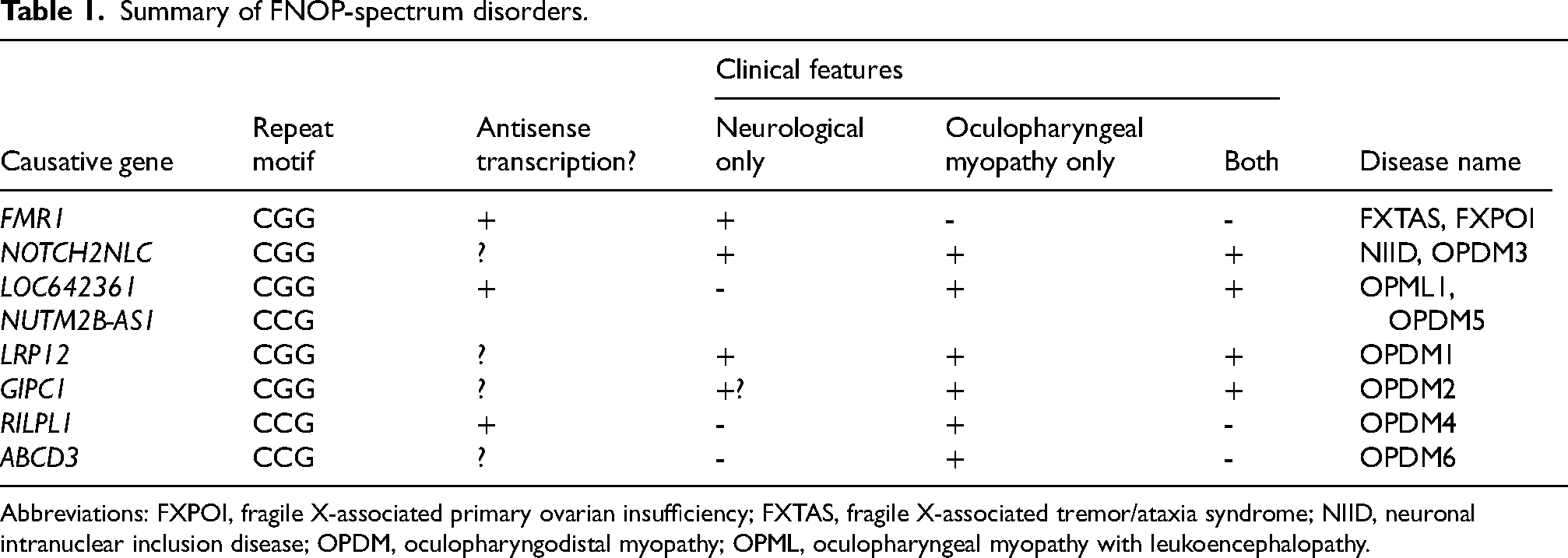
Abbreviations: FXPOI, fragile X-associated primary ovarian insufficiency; FXTAS, fragile X-associated tremor/ataxia syndrome; NIID, neuronal intranuclear inclusion disease; OPDM, oculopharyngodistal myopathy; OPML, oculopharyngeal myopathy with leukoencephalopathy.
From the perspective of the repeat motif–phenotype correlation, in which similar repeat sequences in several genes cause similar diseases, the pathogenesis of OPDM can be broadly divided into RNA toxicity or the toxic effect of repeat-associated non-AUG-initiated (RAN) translation products. Little is known about why expansions of CGG and CCG repeats cause similar diseases. Since antisense transcription commonly occur in repeat expansion diseases, this may be the mechanism by which CGG and CCG repeats cause similar diseases, although this is not yet extensively examined as discussed before (Table 1).
If molecules that bind both CGG and CCG repeat RNAs exist, however, sequestration of such proteins to RNA foci may underlie the common pathogenesis (Figure 2(a)). Such molecules would be good therapeutic targets that could be used commonly in many OPDM patients. Another possibility is RAN translation (Figure 2(b)). The pathomechanism of RAN translation is discussed in the next section.
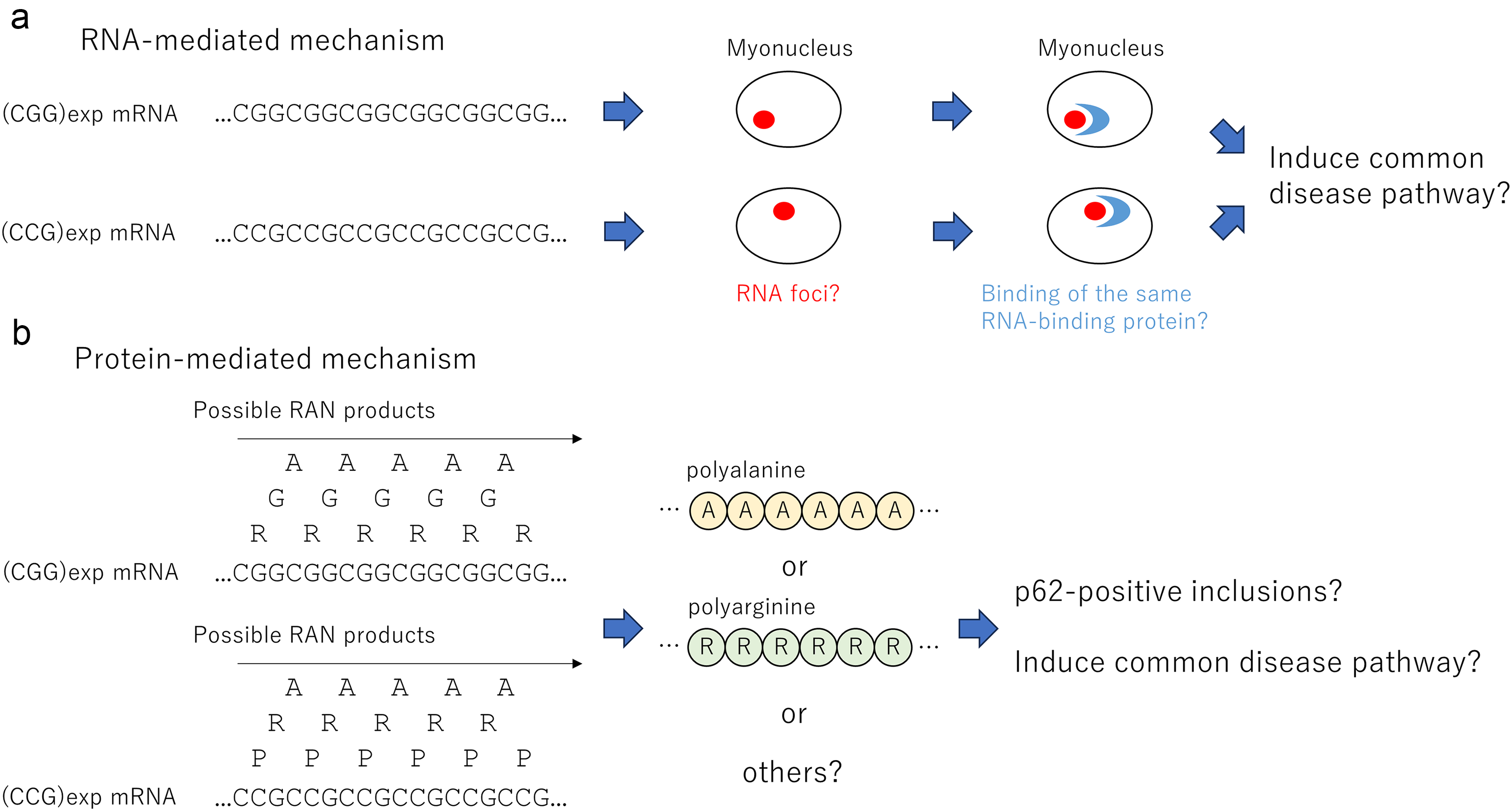
Possible pathomechanisms caused by expansions of CGG and CCG repeats. (a) OPDM is caused by expansions of CGG and CCG repeats. The reason is unclear at this time. In this figure, the RNA-mediated mechanism is assumed. It is possible that repeat RNAs produced by expanded CGG or CCG repeats cause RNA foci in myonuclei, which sequester the same RNA-binding proteins. (b) In this figure, a common disease pathway caused by RAN translation is assumed. Theoretically, polyalanine, polyglycine, and polyarginine are produced by expansions of CGG repeats, whereas polyalanine, polyarginine, and polyproline are produced by expanded CCG repeats. It is possible that commonly produced polyalanine or polyarginine proteins constitute p62-positive inclusions and induce a common disease pathway.
Is it polyalanine, polyglycine, or something else?
The phenomenon of RAN translation is widely known in neuromuscular diseases caused by repeat expansion mutations. In these diseases, translation begins in the absence of an AUG codon, presumably related to changes in the three-dimensional structure of mRNA due to the presence of repeat sequences.
For the same CGG repeat disease, FXTAS, RAN translation has been shown to occur. Translation is initiated from an AUG-like sequence upstream of the repeat called the near-cognate sequence, and proteins, particularly those in the polyglycine frame, are considered to be involved in the pathogenesis.45,46,47 For NIID, there is an AUG codon upstream of the CGG repeat, and expanded repeats have been shown to initiate translation from this AUG codon in the polyglycine frame.48,49
At present, the RAN translation of OPDM has not yet been fully clarified. The formation of p62-positive inclusions in muscles of OPDM patients suggests the production of aberrant proteins. Only a conference paper shows the in vitro production of RAN proteins in polyglycine frame from CGG repeat expansions in a LRP12 construct. Thus, like FXTAS and NIID in FNOP-spectrum disorders, OPDM may be classified into a polyglycine disease.
Another thing to consider is the recently discovered polyglycine repeat expansions in ZFHX3 in patients with spinocerebellar ataxia type 4 (SCA4).50,51,52,53 SCA4 is the first disease found to be caused by polyglycine repeat expansions in coding regions. SCA4 does not exhibit high-intensity signals on DWI or in the middle cerebellar peduncles, which are typically observed in conditions like FXTAS and NIID. However, it presents with sensory neuropathy, autonomic dysfunction, oculomotor abnormalities, and dysphagia. It is intriguing that some of these clinical features overlap with those seen in FXTAS and NIID, suggesting potential similarities in their clinical manifestations despite differences in imaging findings.
On the other hand, if the expansions of CGG repeats result in RAN translation, polyalanine, polyglycine, and polyarginine will be produced. Expansions of CCG repeats will produce polyalanine, polyarginine, and polyproline. The fact that expanded CGG and CCG repeats cause similar diseases may suggest a common pathogenic mechanism involving RAN proteins, particularly polyalanine and polyarginine proteins. The polyalanine sequence is of particular interest, considering the expanded polyalanine stretch in PABPN1 in OPMD. Since the repeat lengths observed in OPDM and OPMD (>50 repeat units mainly in noncoding regions and 11–18 polyalanine stretch, respectively) and the diameters of filamentous inclusions revealed by electron microscopy (10–20 nm and 8.5 nm, respectively) are quite different, the underlying mechanisms may be complex. In a mouse model of NOTCH2NLC-related disease, inclusions were found containing NOTCH2NLC-polyglycine, as well as NOTCH2NLC-polyalanine and NOTCH2NLC-polyarginine proteins. Furthermore, hnRNPM was shown to interact with both NOTCH2NLC-polyglycine and NOTCH2NLC-polyalanine. 54 Further studies are needed to determine whether a correlation exists between RAN proteins and disease phenotype.
Summary
In this paper, an overview of OPDM is discussed. Since the disease concept was published in 1977 and the causative mutations were finally identified in 2019, much has become known about OPDM over the past five years. It is our strong hope that the pathogenesis of this rare disease, for which there is no cure, will be further elucidated and that appropriate treatments will be developed.
Footnotes
Funding
The author received grants from the Japan Agency for Medical Research and Development (AMED) under Grant Number JP23ek0109673 and from JSPS KAKENHI under Grant Number JP23K27514.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.