
Abstract
Skeletal muscle relies on its inherent self-repair ability to withstand continuous mechanical damage. Myofiber-intrinsic processes facilitate the repair of damage to sarcolemma and sarcomeres, but it is the coordinated interaction between muscle-resident satellite and stromal cells that are crucial in the regeneration of muscles to replace the lost muscle fibers. Fibroadipogenic progenitors (FAPs), are muscle-resident mesenchymal cells that are notable for their role in creating the dynamic stromal niche required to support long-term muscle homeostasis and regeneration. While FAP-mediated extracellular matrix formation and the establishment of a homeostatic muscle niche are essential for maintaining muscle health, excessive accumulation of FAPs and their aberrant differentiation leads to the fibrofatty degeneration that is a hallmark of myopathies and muscular dystrophies. Recent advancements, including single-cell RNA sequencing and in vivo analysis of FAPs, are providing deeper insights into the functions and specialization of FAPs, shedding light on their roles in both health and disease. This review will explore the above insights, discussing how FAP dysregulation contributes to muscle diseases. It will offer a concise overview of potential therapeutic interventions targeting FAPs to restore disrupted interactions among FAPs and muscle-resident cells, ultimately addressing degenerative muscle loss in neuromuscular diseases.
Keywords
Introduction
Skeletal muscle has a remarkable ability to repair from injury, enabling it to maintain tissue health in the face of mechanical damage caused by strenuous physical activity, trauma, and diseases. Injured muscle fibers efficiently repair damage to their plasma membrane, but if repair fails the muscle fiber undergoes necrosis. 1 It is then fully regenerated by dedicated stem cells, known as muscle satellite cells (MuSCs).2,3 While plasma membrane repair is a myofiber-intrinsic response, regeneration is a myofiber-extrinsic process involving coordination between MuSCs and other muscle-resident cell types.1,4–6 During regenerative myogenesis MuSCs proliferate, differentiate, and then fuse to regenerate the necrotic myofibers. 2 The combination of inflammatory cells and muscle-resident mesenchymal progenitors, called fibro/adipogenic progenitors (FAPs), form the stromal niche that supports regenerative myogenesis by the MuSCs (Figure 1). Although muscle stromal cells don’t directly form myofibers, they aid regeneration by clearing necrotic debris, producing extracellular matrix (ECM), and releasing factors that support the maintenance, activation, and fusion of muscle stem cells (MuSCs), thus their depletion impairs myofiber regeneration.5,7–9
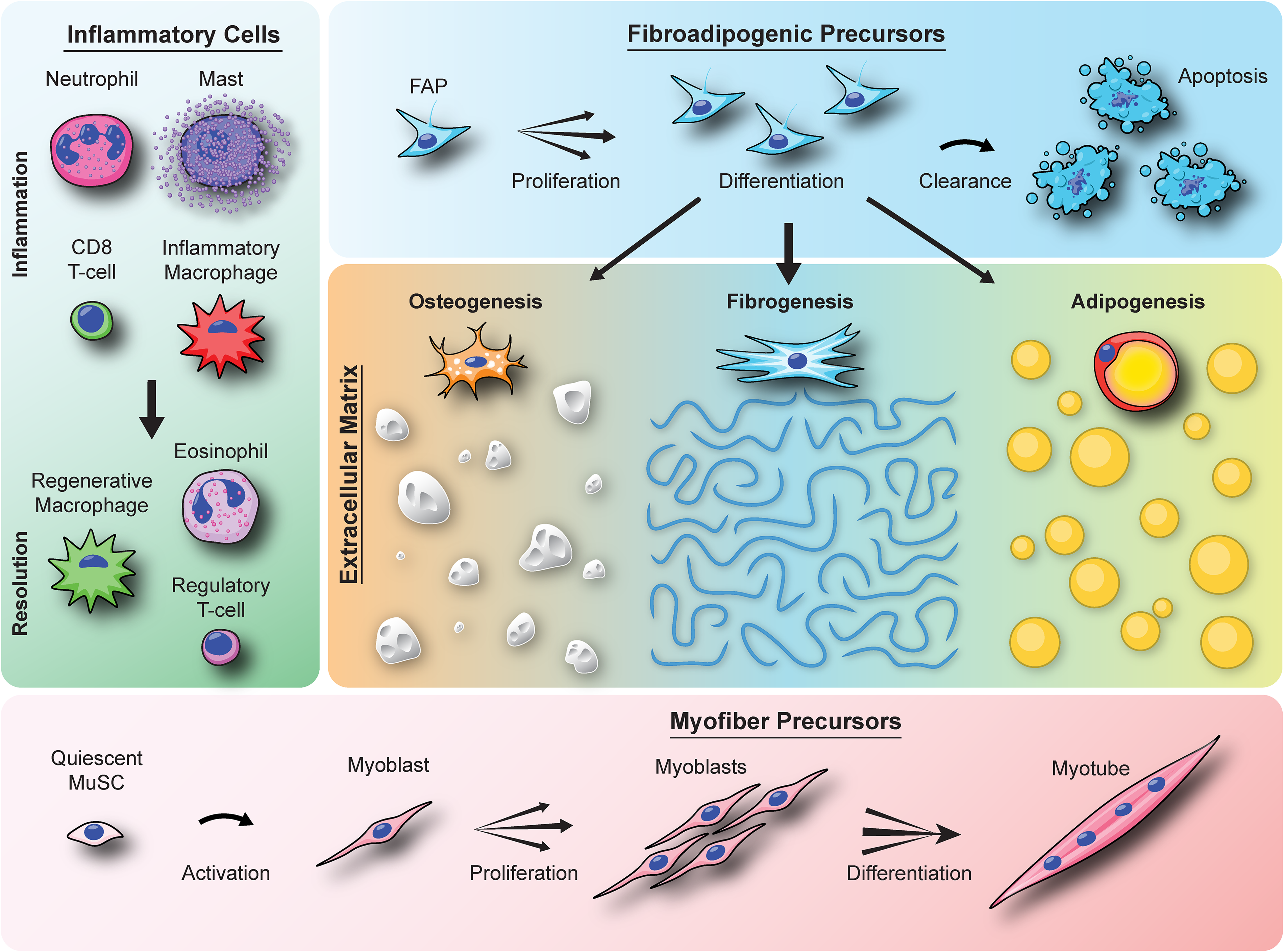
Illustration of the cellular players that drive muscle re/degeneration. The schematic shows the three classes of muscle-resident mononuclear cells that play a part in the repair or loss of injured skeletal muscle. These include the inflammatory cells that migrate from blood to the site of injury to initiate the repair process, the MuSCs that undergo myogenic fusion to form the multinucleated myofibers, and the FAPs that proliferate and differentiate to form the ECM which supports the regenerated myofibers. However, an aberrant inflammatory or myogenic responses in diseases can trigger the FAPs to adopt an alternate differentiation program including osteogenesis, fibrogenesis, and/or adipogenesis that eventually lead to degenerative tissue matrix and loss of muscle function.
Muscle damage triggers the circulating inflammatory cells to enter the injured muscle and initiate the regeneration process, where they interact with and trigger the proliferation of the FAPs.4,7,10 FAPs are marked by Platelet-derived growth factor receptor alpha (PDGFRɑ), with over 70% of FAPs expressing this marker in mouse and human muscle.11–13 FAPs can also be identified in mice as muscle-resident cells expressing the stem cell antigen 1 (Sca1), but lacking the expression of hematopoietic, endothelial, and satellite cell markers (SCA1+/CD31−/CD45−/α7-integrin−), 14 or in human muscle by expression of CD15 or CD34 in combination with negative selection for other lineages.15,16 Single cell RNA-Seq analysis of muscles shows that either of the mouse FAP selection criteria results in populations that are nearly indistinguishable from each other by gene expression, but notably distinct from MuSCs. 11 Further, unlike MuSCs, which are found inside the myofiber basal lamina, FAPs are present outside the basal lamina, and closer to the blood vessels. 17 FAPs are present at low levels in healthy muscles, but rapidly proliferate upon muscle injury and appear to facilitate regeneration, before being cleared by inflammation-induced apoptosis as the muscle returns to homeostasis (Figure 1). 18 In chronic muscle diseases, however, clearance of FAPs is impaired, which causes their increased accumulation in the ECM.19,20 FAPs are mesenchymal progenitors, and hence possess fibrogenic, adipogenic, osteogenic, and chondrogenic differentiation potential.10,12,14,19 Consequently, FAPs that escape clearance, accumulate in diseased muscle and undergo differentiation leading to fibrotic, adipogenic or calcified deposits that gradually replace the adjacent myofibers - a pathology shared across numerous neuromuscular diseases (Figure 1).19–21 Due to these common features of FAPs across different muscle diseases, preventing their aberrant accumulation and differentiation can offer promising therapeutic targets. 22 However, this approach needs to be carefully balanced as a complete lack of FAPs causes myogenic deficits, limiting the clinical potential of this strategy in dystrophic muscle.7,9 Below we will discuss the current understanding of the role of FAPs in healthy and diseased muscles, with a focus on disease etiology and efforts to harness this for therapy.
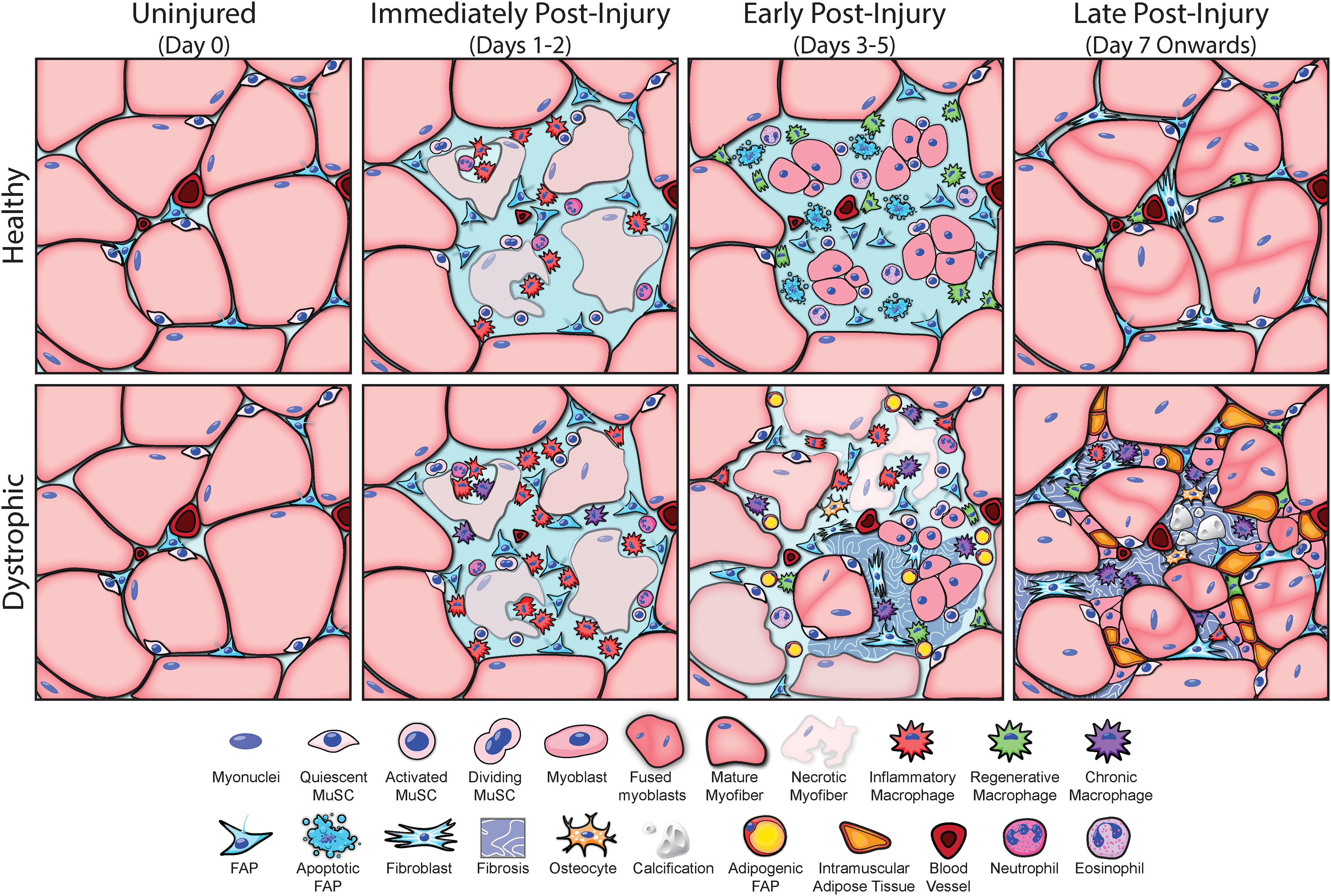
Cellular choreography during acute repair stages of injured healthy and dystrophic muscle. The schematic shows the contrast between the well-coordinated regenerative response following a bout of acute injury in healthy muscle vs dysregulation of this process in dystrophic muscle. Muscle regeneration is a highly dynamic process, and the static images here represent events starting prior to onset of dystrophic symptoms or any muscle injury and in the days following injury grouped into immediately (1–2 days), early (3–5 days), and late (beyond 7 days) post injury. In healthy muscle, inflammatory cells enter the site rapidly after myofiber injury to clear debris from damaged myofibers and the basement membrane. During the early stages of repair, the injury site becomes inflamed and supports MuSC and FAPs proliferation and onset of their differentiation to begin reforming the lost myofiber and basement membrane respectively. The niche then transitions over the next 1–2 weeks to complete regeneration of the lost myofiber and clearance of FAPs to return the muscle to its homeostatic state. In dystrophic muscle, this process is dysregulated leading to incomplete repair which is compounded by subsequent rounds of damage and regeneration deficit that causes chronic activation of macrophages and failure of the injury site to sequentially transition from an inflammatory to a resolving niche. This loss of cellular choreography and associated cellular cues cause FAPs to proliferate excessively and accumulate in the muscle by failed clearance and begin their differentiation into fibroblasts or adipocytes which replaces the lost myofibers with fibrotic or adipogenic deposits.
Association between FAPs and muscle health
FAPs (and their precursors) play a fundamental role in the development and homeostatic maintenance of skeletal muscle. Non-myogenic mesenchymal progenitors have been identified as essential to the development of skeletal muscle as they provide the required cues for correct patterning of myofiber orientation and fiber type. 23 These cells are identified by expression of transcription factor 4 (TCF4; transcription factor 7-like 2; Tcf7l2), 24 which is also expressed in FAPs found in adult muscles. 25 Lineage tracing in transgenic mice shows that a population of these embryonic TCF4+ mesenchymal cells give rise to mature muscle-resident FAPs,26,27 suggesting that FAPs play a continued role in tissue maintenance. Consistent with this idea, chronic depletion of PDGFRa+ cells in transgenic mice results in loss of muscle mass, force production, and a reduced MuSC pool. 7 FAPs are also intrinsic to muscle regeneration post-injury; this is evident from the rapid increase in FAP numbers triggered by muscle injury, which peaks by 3–5 days after healthy muscle damage and coincides with myogenic fusion4,14 (Figure 2). Co-culture with FAPs enhances myoblast differentiation and fusion, and there is an age-dependent loss of this pro-myogenic ability of FAPs due to reduced matricellular signaling through factors secreted by the FAPs.8,14 Although, not formally linked, the secretion of pro-myogenic factors is thought to underpin the role of FAPs in both long-term maintenance of muscle and acute regeneration of the tissue after damage.
Cell intrinsic and extrinsic regulators
Insights into how FAP-intrinsic responses can help maintain various aspects of muscle health are being offered by recent studies using single cell RNA-Seq. These studies are revealing a diversity of FAP states based on their transcriptomic profiles.11,16,28–33 At rest, FAPs are enriched in ECM formation and organization genes such as Gsn, Dcn, Adamts5 and Dpep1, but not immune or inflammatory genes.11,28 Two days after muscle injury, FAPs expressing chemokines such as Ccl7 and Cxcl1 increase, followed by FAPs enriched in ECM synthesis genes like biglycan and collagens at day 5, before returning to the uninjured profile by day 7. 28 These intrinsic changes in FAP behavior in response to the tissue environment are consistent with them playing an active role after muscle injury. Different FAP subpopulations have also been confirmed via studies in transgenic mice; hypermethylated in cancer 1 (Hic1) marks a quiescent FAP-like population, and lineage tracing shows Hic1+ cells give rise to multiple lineages after muscle injury, including fibroblasts, adipocytes and tenocytes. 24 Alternatively, lineage tracing of GLI family zinc finger 1 (Gli1) in FAPs shows that Gli1 marks a minor subpopulation population with increased hedhehog (Hh) signaling at rest, but these rapidly grow to a major one by 3 days after muscle injury. 34 The Gli1+ FAPs show reduced adipogenic potential and increased expression of pro-myogenic factors, which is consistent with other studies which show Hh activation is essential to suppress adipogenesis during muscle regeneration.35,36 In human muscle, a FAP population marked by MME expression show a bias towards adipogenesis, 37 but in contrast, those marked by Thy1/CD90+ appear biased towards fibrogenesis. 16 These different subpopulations are often associated with specific cellular niches, either by their location, or the damage/regeneration state of the muscle, indicating dynamic FAP states in response to their surrounding environment. The effect of the extracellular niche on FAP fate has also been shown using the differences between experimental models of muscle injury; myotoxic injury promotes FAP fibrogenesis and glycerol injury promotes FAP adipogenesis, but cross-transplantation of FAPs between these muscles causes FAPs to adopt the fate that matches the recipient (rather than the donor) tissue niche. 12
In addition to the above responses, FAPs also contribute to their surrounding environment via modulating inflammation, ECM remodeling, and supporting myogenic as well as vasculogenic activities, lack of which impairs muscle health.7,38 In injured muscles, FAP-derived signals such as Bone morphogenetic protein-3B (BMP3B) and interleukin-6 (IL-6), effect MuSC-mediated muscle hypertrophy.9,14,39–42 FAPs also secrete vascular endothelial growth factor (VEGFA), and this seems to have critical effects on muscle and vascular regeneration after ischemic damage. 38 The pro-myogenic effects of FAPs have also been demonstrated in vitro where co-culture with FAPs enhances MuSC fusion, even when physically separated from FAPs, indicating regulation by factors secreted by FAPs. 9 This cellular crosstalk is bidirectional; with co-culture of differentiating MuSCs regulating FAPs, such as blocking their adipogenic differentiation. 12 Further, direct myogenic inhibition via myostatin or ablation of MuSCs is both linked to FAP fibrogenesis and muscle fibrosis,39,43 suggesting that, similar to the in vitro co-culture studies, FAP differentiation during regeneration is regulated to some degree by myogenesis. The muscle niche, in turn, regulates FAPs by contributing to altered cell fates with aging; FAP cultures from aged muscle show enhanced fibrogenic differentiation and reduced ability to support MuSCs, but transplantation of FAPs from young muscle can enhance myogenic commitment of aged MuSCs by paracrine signaling. 8
Inflammatory cells and FAP crosstalk
Inflammatory cells are the other key component of the muscle niche that supports regeneration (Figure 1). In healthy muscle, a subpopulation of FAPs expressing Dpp4 appear to be responsible for maintaining the pool of tissue-resident macrophages via secretion of colony-stimulating factor 1 to promote self-renewal. 44 After muscle injury, neutrophils and mast cells arrive at the site of damage and recruit monocytes to clear the debris, initiating the inflammatory niche.4,45,46 Monocytes which enter injured muscle undergo differentiation into pro-inflammatory macrophages and release TNFα, IL-1β, among other cytokines that activate subsequent inflammatory cascade and trigger proliferation of MuSCs and FAPs.4,18,45,47 Through phagocytosis and efferocytosis, these macrophages clear debris and activate transition to the pro-regenerative phase of inflammation. The macrophages then secrete cytokines such as TGF-β and IL-10 which promote myoblast fusion and activate FAPs to support ECM remodeling.18,45–47 Knockout of Ccr2 prevents macrophage infiltration into the muscle; this impairs myofiber regeneration and leads to fibrofatty expansion of the ECM after muscle injury,48,49 which suggests FAP dysfunction in the absence of typical inflammatory cues.
Eosinophils also arrive in the acute inflammatory phase (Figure 1). These cells secrete interleukins (IL-4, IL-13) and activate FAPs expressing the IL-4 receptor, causing them to proliferate and this can be recapitulated in vitro. 49 Further, use of conditioned media from IL-4-activated FAPs induces myoblast differentiation. 49 Steroid inhibition of IL-4 also increases FAP adipogenesis in injured muscles and this is recapitulated in the injured muscle of IL-4-knockout mice. 50 Inflammatory cues such as macrophage-derived TNFα triggers FAP apoptosis, enabling clearance of the FAPs accumulated in the injured tissue and helping FAPs to return to pre-injury levels. 18 This transition can be inhibited by a niche that is high in the levels of TGF-β, which is produced by the pro-regenerative and pro-fibrotic macrophages.18,47 Thus, FAP abundance and fate choice are both influenced by macrophage-generated signals in the muscle niche. In support of this mode of action of macrophages, in vitro use of macrophages polarized using IL-1β versus IL-4 promote different FAP fates such that IL-1β-treated macrophages reduce FAP adipogenesis, while IL-4-treated macrophages enhance FAP adipogenesis. 51 The opposing effects of IL-4-treated macrophage signaling versus direct IL-4 treatment on FAP adipogenesis during healthy muscle regeneration highlight the complexity of the niche and the relative timing of macrophage-FAP interactions in the FAP response during healthy muscle regeneration. As FAPs also secrete signals that modulate invading inflammatory cells, which in turn regulate FAPs, this creates the triumvirate of inflammatory cell-FAP-MuSC interactions that are crucial for healthy muscle repair, disruption of which can result in muscle diseases (Figure 2).
Association between FAP fate and muscle disease
As described above FAPs can adopt osteogenic, fibrogenic or adipogenic fates (Figure 1). A complex cellular choreography between FAPs, inflammatory cells, and MuSCs maintains muscle homeostasis by acutely responding to physiological muscle damage (Figure 2). However, muscle diseases exhibit chronic muscle damage, both due to and leading to the dysregulation of these interactions. These eventually manifest as tissue degeneration due to progressive replacement of functional myofibers with non-contractile ECM,52,53 which is created by FAP differentiation (Figure 2). As progressive fibroadipogenic ECM expansion correlates with clinical severity of disease, and FAPs are the cellular origin of this ECM expansion, targeting FAPs is a promising therapeutic strategy for a wide range of neuromuscular disorders.22,54
Adipogenesis
Excessive FAP accumulation drives fatty replacement of muscles in patient and mouse models of dysferlinopathy – Limb Girdle Muscular Dystrophy R2 (LGMDR2). 19 MRI studies have confirmed that fatty replacement of muscle is strongly correlated with functional decline in these patients. 55 The consistency of these findings highlights the clinical importance of FAP dysregulation in dysferlinopathy and identify it as a therapeutic target. However, FAP-driven fatty replacement is not limited to dysferlinopathy; facioscapulohumeral dystrophy (FSHD) also shows fatty infiltration in the muscle by MRI, which correlates with increased accumulation of FAPs and histopathological features.55,56 FAP-driven intramuscular adipogenesis is also not limited to dystrophies, as it is a common feature of muscles that poorly regenerate after rotator cuff tears and in sarcopenia.57,58
Fibrogenesis
FAP dysfunction is also described in Duchenne muscular dystrophy (DMD); again, FAP accumulation is a prominent feature of DMD patient muscle biopsies and mouse models, but (in contrast to LGMDR2) this correlates with an increase in muscle fibrosis. 59 Interestingly, FAP dysfunction in FSHD patients has also been associated with interstitial fibrosis, 56 which is in addition to the increase in adipogenesis noted above. FAPs have also been implicated in the fibrosis seen in biopsies of affected muscles in oculopharyngeal muscular dystrophy patients and human muscles which naturally feature ECM deposition with age. 60 FAP-driven muscle fibrosis is also not limited to dystrophies; dysregulation of collagen expression has been reported in FAPs from inclusion body myositis, 32 an inflammatory myopathy which features fibrotic expansion of the ECM in certain muscles.61,62 Similar features - FAP accumulation and muscle fibrosis, are also observed in diseases which feature denervation of the muscle, including amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA).63,64
Osteogenesis
Because FAPs are tissue-resident mesenchymal stromal cells that possess osteogenic and chondrogenic potentials, in addition to their fibroadipogenic properties, these cells can also mediate heterotopic ossification in muscle. 65 Dysregulation of activin signaling in FAPs causes Fibrodysplasia Ossificans Progressiva (FOP), a debilitating musculoskeletal disorder. 66 Interestingly, FAP dysregulation also leads to ectopic calcification in muscles of a severe DMD mouse model - D2-mdx. 21 Calcium deposits have been observed in biopsies from DMD and, to a lesser extent, myotonic dystrophy patients, 67 suggesting osteogenesis as a further mechanism by which FAP dysfunction can contribute to the pathogenesis of chronic muscle diseases.
Insights into diseased muscle niche
The conditions described above vary considerably in their etiology but each of them manifest FAP dysregulation in the muscle. Rather than specific alterations in FAPs for each disease that produce converging phenotypes, these commonalities suggest that chronic muscle damage creates a dysregulated muscle niche that leads to FAP dysfunction, committing them to aberrant cell fates. In support of this idea, freshly isolated FAPs from dysferlinopathic muscle exhibit spontaneous adipogenesis when cultured ex vivo, but FAPs from WT do not and instead require exogenous induction to show similar adipogenic differentiation. 19 This indicates that a bias towards FAP adipogenesis is generated by the extracellular niche in dysferlinopathic muscle, which is retained by the freshly isolated cells. ScRNA-Seq analysis also pointed to the importance of the niche in their fate, as PDGFRα+ FAPs isolated from wildtype and dysferlinopathic muscles show significant transcriptional similarities, but with reduced expression of adipogenic repressors in the dysferlinopathic FAPs. 11 Single-cell analysis of the mdx mouse model of DMD also demonstrates broad transcriptional similarities between WT and dystrophic FAPs, but mdx FAPs show closer expression profiles to those from injured WT muscle. 41 Given the persistent damage and regeneration in DMD muscle this again points to niche-driven dysregulation of FAPs in dystrophic muscle rather than intrinsic alterations to FAP behavior resulting from the lack of either dystrophin or dysferlin. 68
Incomplete transition from the early pro-inflammatory state to the pro-resolving state due to chronic and asynchronous muscle damage is a hallmark of the dystrophic muscle niche, which drives fibroadipogenic degeneration of the muscle.47,68 Lack of the coordinated inflammatory response required for successful muscle regeneration drives pathology in dystrophic muscles. Dysferlin-deficient macrophages exhibit altered phagocytosis, efferocytosis, and even and cyto-destructive ability,69,70 which change the niche after muscle damage. Dystrophin-deficient muscle also accumulates immune cells including pathogenic macrophages and eosinophils among others, that combine to create a niche that promotes increased accumulation and fibrotic differentiation of the FAPs18,59,71–74 . ScRNAseq and spatial transcriptomic analysis of D2-mdx muscle, showed that proinflammatory signals generated at recently damaged areas mix with the profibrotic signals in regenerating areas that contributes to widespread in situ changes in muscle gene expression, which leads to the severe muscle pathology. 75 In addition to the dysregulated niche, use of scRNAseq analysis also shows that dystrophic muscles are enriched in a population of pathogenic macrophages marked by high expression of galectin-3 and osteopontin (Spp1). 71 Increased osteopontin enhances local TGF-β activation through matrix metalloprotease (MMP) 9 and is linked to fibrosis. 76 It also alters FAP-macrophage interactions in a manner that promotes fibrosis. 71 While the precise mechanisms driving FAPs to a matrix-depositing fate are not well characterized, the canonical Wnt pathway has been implicated in mdx muscle. 77 To further elucidate these cellular interactions, a recent study reported a model whereby FAPs and macrophages secrete distinct subunits of the complement C1 for combinatorial activation of pro-fibrotic Wnt signaling. 78 These findings illustrate the critical role that inflammatory niche dysregulation plays in driving pathogenic FAP differentiation in chronically damaged muscle.
Therapeutic strategies to correct FAP dysfunction in muscle diseases
Therapeutic approaches for treating degenerative muscle diseases have largely focused on countering chronic inflammation via the use of glucocorticoids. This strategy delays degeneration and promotes muscle function, but it is also associated with adverse effects such as induction of adipogenic differentiation by inhibiting IL-4 signaling. 50 Efforts to develop anti-inflammatory therapies that do not involve steroids have led to testing metformin - an FDA-approved AMPK activator with potential for addressing metabolic dysfunction. 79 Metformin's effects on mitochondrial biogenesis in preclinical studies have been linked to reduced fibrosis without the side effects associated with corticosteroids; however, its effectivity required supplementation with L-arginine or L-citrulline, and the benefits are not evident in clinical trials.79,80 Another therapeutic direction is the use of pro-resolving bioactive lipids, such as resolvins, which show pre-clinical evidence of promoting resolution of inflammation and improving myogenesis.81,82 However, neither drug directly suppress the accumulation or aberrant differentiation of FAPs, so efforts to further develop such inflammation-targeting therapies are needed.
Aside from targeting inflammation, other ongoing efforts to therapeutically target FAPs have focused on the prominent pathological aspects of FAPs– excess accumulation and aberrant differentiation, and have yielded mixed success in preclinical studies. PDGF signaling induces FAP proliferation 83 and TGF-β inhibits FAP clearance. 18 As the receptors for both PDGF and TGF-β function via tyrosine kinases, tyrosine kinase inhibitors (TKIs) were among the first molecules tested to prevent FAP accumulation in the diseased muscle. These drugs, including Imatinib,84–86 Nintedanib, 87 Nilotinib, 18 and Crenolanib, 88 show similar benefits when tested in mdx mice. They lead to decreased FAP accumulation in dystrophic mdx muscle which correlates to less fibrosis. The dual inhibition of PDGFR and TGF-β signaling by TKIs makes it difficult to identify the precise mechanism of action which produces these effects, but it should be noted that direct manipulation of PDGFRα splicing to prevent signaling also prevents FAP accumulation and ECM deposition after healthy muscle injury. 83 This suggests that PDGFRα inhibition likely contributes to the efficacy of TKIs in reducing FAP accumulation and fibrosis. Despite this promise, TKIs also result in myogenic deficits after acute muscle injury 89 which mimic some of those observed with FAP ablation. Because muscular dystrophy patients will require chronic treatment across their lifespan, the consistent association between FAP depletion and defective myogenesis suggests that TKIs and other similar approaches to clear FAPs from the tissue are unlikely to be useful in long-term treatment of muscle loss and weakness seen in these disorders.
This leaves targeting aberrant FAP differentiation as the alternative approach for therapeutic intervention, with several studies showing the promise of such agents using preclinical models. Ciliary Hh signaling has been shown to mediate physiological repression of FAP adipogenesis via inhibition of MMP-14 35 and mimicking this with the MMP inhibitor batimastat, prevents adipogenic replacement of muscle after glycerol-induced injury and in dysferlinopathy.19,35 Interestingly, FAP-derived MMP-14 has also been implicated in the activation of latent TGF-β in the ECM, which is associated with fibrosis in dystrophic muscle. As a result, MMP-14 inhibition also reduces collagen deposition after fibrotic injury in mdx mice, making this mechanism a possible panacea for ECM fibroadipogenesis in dystrophic muscle. MMP inhibitors were originally developed to treat cancer, but clinical trials were universally unsuccessful due to a lack of efficacy and significant side effects, including a musculoskeletal syndrome which affected most patients on the drug.90,91 There are ongoing efforts to reduce the side-effect profile of MMP inhibitors by improving target selectivity, 92 but it remains to be seen if an efficacious inhibitor of MMP-14 amenable to long-term chronic treatment can be developed.
As discussed above, freshly isolated FAPs from dystrophic muscle retain the properties of their niche and will spontaneously differentiate when cultured in vitro. This suggests the possibility of epigenetic changes conferred by the dystrophic niche which alters the cell fate decisions of FAPs from diseased muscle. Consistent with this hypothesis, histone deacetylase inhibitors (HDACi) have shown promising results to manipulate FAP trajectories in dystrophic muscle. Treatment with Givinostat restricts the development of fibrosis in both mdx muscle93,94 and DMD patient biopsies. 95 Adipogenic replacement is also ameliorated in givinostat-treated DMD patients, and givinostat blocks mdx FAP adipogenesis in vitro. 96 These suggest that HDACi is a promising approach to block fibroadiopgenesis in dystrophic muscle. An alternative HDACi, trichostatin A has also been shown to restrict fibrosis in mdx muscle 97 and block adipogenic replacement after rotator cuff tears 98 which further highlights the potential of this pathway in preventing ECM expansion in degenerating muscle. Interestingly, preventing fibroadipogenesis via HDACi has the additional benefit of improved myogenesis by altering the secretory profile of dystrophic FAPs.99,100 However, optimism for this approach is tempered by the observations that FAPs from aged dystrophic muscle are largely resistant to the effects of HDACi,96,101 casting some doubt on their efficacy as a chronic treatment strategy.
More recently, the Wnt/β-catenin signaling pathway has been shown as a potential mechanism to arrest FAP adipogenesis. Cellular β-catenin is degraded as part of the induction of adipogenensis, and stabilizing β-catenin by inhibiting glycogen synthase kinase 3b (GSK3B) inhibits FAP adipogenesis in the mdx model of DMD. 102 Similarly, pharmacological activation of the Wnt pathway also suppresses FAP adipogenesis and intramuscular lipid deposition after rotator cuff tears in mice. 103 Collectively, these results suggest a potential mechanism of regulating FAP adipogenesis via raising β-catenin levels. However, a recent report shows that increased β-catenin can cause FAP activation without the trigger of muscle injury and canonical activation of the Wnt pathway in FAPs long-term leads to muscle atrophy and fibrosis. 104 These findings suggest that manipulating canonical Wnt pathways to suppress FAP adipogenesis is likely to be associated with significant side effects, which may preclude their use in dystrophic patients. A specific Wnt ligand (Wnt7a) has been shown to suppress FAP adipogenesis without raising β-catenin, 105 which preserves some hope that non-canonical Wnt pathways may be a useful tool in suppressing FAP adipogenesis.
As we allude to above, the multipotency of FAPs creates challenges in FAP-targeting therapies as the fibrogenic and adipogenic cell fates are often in competition and inhibition of one fate can lead to increases in the other. For example, in vitro treatment of FAPs with IL-15 enhances expression of desert hedgehog (DHH) and Timp3, directly preventing their adipogenic differentiation in vitro and in injured muscle in vivo. 106 However, extended use of IL-15 causes increased fibrosis and as such, IL-15 enriched areas of the muscle show increased accumulation of FAPs and fibrotic collagen after rotator cuff tears. 106 To avoid side-effect profiles which preclude clinical use, it is necessary to have a wide range of FAP-targeting agents which can be tested across different situations. To identify novel modulators of FAP fate, a popular strategy is to induce fibro adipogenic differentiation of FAPs in vitro and screen for small molecules which inhibit the process. This has been used as a high throughput screening strategy to identify inhibitors of FAP fibroadipogenesis, leading to the identification of promethazine, 107 azathioprine 108 and GSK3B inhibitors 102 as drugs that restrict FAP adipogenesis. With the MSC lineage of FAPs, the method to induce MSC adipogenesis in vitro is also efficacious for FAPs. Thus, it is likely that drugs shown to restrict MSC adipogenesis in vitro will also show efficacy against FAPs in this assay. This potential is demonstrated by several drugs including Epigallocatechin-3-gallate, 109 Flufenamic Acid, 110 and Chidamide. 111 However, there is a disconnect between the exogenous induction of FAP differentiation in vitro and the dystrophic niche that endogenously drives the process in vivo. As a result, not all drugs discovered using in vitro screens will be useful for the treatment of dystrophies. This conundrum has been illustrated by a high-throughput screen which identified multiple inhibitors to prevent induced mdx FAP fibrogenesis in vitro, but none of these molecules reduced fibrosis in mdx muscle in vivo. 54 Thus, there remains an unmet need to identify drug therapies that can efficiently target FAP fate dysregulation in vivo in a manner such that their use in vivo will address the various pathological potentials of FAPs in diseased muscles.
Based on the therapeutic approaches discussed for targeting FAPs, several strategies for improving muscle health have emerged. These include -
Lessons learnt and future directions
Research on FAPs has significantly advanced our understanding of their biology and function in muscle health, highlighting their crucial role as regulators of skeletal muscle homeostasis and regeneration. However, gaps in our knowledge still exist regarding the specialization of FAPs, their interaction with other cell populations and the ECM in their niche, and how they govern their differentiation paths in muscle. While FAPs are essential for tissue repair post-acute injury, uncontrolled proliferation results in fibroadipogenic degeneration. It is therefore imperative to explore the molecular switch for FAP differentiation and develop strategies to prevent fibrogenesis and adipogenesis. Such tools would have the potential to both prevent ECM expansion and enhance myogenesis by rebalancing FAPs towards their homeostatic function in healthy muscle. This understanding is crucial for regulating FAP fate and molecular interactions, but the heterogeneity of FAPs and complexity of their cellular niche pose challenges in assessing their function in quiescent versus acutely and chronically damaged muscle tissue. Examining how growth factors and proteins derived from FAPs contribute to muscle degeneration and therefore the viability of targeting them clinically to alleviate muscle atrophy is also essential. FAP-targeting therapies should correct FAP dysregulation to restrict aberrant accumulation and differentiation. However, achieving this chronically through therapeutic approaches remains elusive due to poor understanding of the molecular underpinnings of FAP proliferation and differentiation in disease-specific context. Deciphering these and the dual role of FAPs requires understanding intrinsic FAP properties and extrinsic factors in the FAP niche that regulate their response to injury and disease. This comprehensive understanding is crucial for advancing therapies aimed at correcting FAP dysregulation and for targeting FAPs as an approach to promote muscle health.
Footnotes
Acknowledgements
Authors acknowledge support of their work by funding from the Jain Foundation and the National Institutes of Health grants 1K01AR077686 and 5R01AR055686.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
MH and JKJ have a pending patent application that relates to the topic of this review, but these and all other authors have no other potential conflicts of interest with respect to the research, authorship, and/or publication of this article.