
Abstract
GNE myopathy is an autosomal recessive hereditary muscle disorder that has the following clinical characteristics: develops in early adulthood, gradually progresses from the distal muscles, and is relatively sparing of quadriceps until the advanced stages of the disease. With further progression, patients become non-ambulatory and need a wheelchair. There is growing concern about extra-muscular presentations such as thrombocytopenia, respiratory dysfunction, and sleep apnea syndrome. Pathologically, rimmed vacuoles and tubulofilamentous inclusions are observed in affected muscles. The cause of the disease is thought to be a sialic acid deficiency due to mutations of the GNE gene required for in vivo sialic acid biosynthesis. Sialic acid supplementation to a presymptomatic GNE myopathy mouse model was effective in preventing the development of the disease. Several clinical studies have been conducted to evaluate the safety and efficacy of sialic acid supplementation in humans. Based on the favorable results of these studies, an extended-release aceneuramic acid formulation was approved for treatment of GNE myopathy in Japan in March 2024. It is anticipated that it will be a significant step in the development of an effective treatment for GNE myopathy and other ultra-orphan diseases.
Overview of GNE myopathy
Introduction
GNE myopathy is a rare genetic disease characterized clinically by a slowly progressive decrease in muscle strength and function, typically beginning with distal parts of the legs.1–3 Pathological features revealed by biopsy of impaired skeletal muscles include rimmed vacuoles and tubulofilamentous inclusions.4–7 In 1981, Nonaka and others in Japan first reported the disease as “distal myopathy with rimmed vacuoles (DMRV)”. In 1984, Argov and Yarom described a similar disease, rimmed vacuole myopathy sparing the quadriceps, which was subsequently referred to as “hereditary inclusion body myopathy (HIBM)”.5,8 In 2001, it was found that both DMRV and HIBM are caused by mutations in the GNE gene involved in sialic acid biosynthesis.6,9–15 Consequently, the name of the disease has been unified under the term “GNE myopathy”. 16
Epidemiology
Patients with GNE myopathy are found frequently in regions from Japan to the Middle East but are distributed worldwide.14,17–24 The Registry of Muscular Dystrophy (REMUDY) for neuromuscular diseases was developed in 2009 by the National Center of Neurology and Psychiatry (NCNP) in Japan.25,26 As of February 2024, 242 Japanese patients have been registered in REMUDY. The international online GNE Myopathy Disease Monitoring Program (GNEM-DMP) was set up in 2012 by a partnership between Translational Research in Europe-Assessment and Treatment of Neuromuscular Disease (TREAT-NMD) and Ultragenyx Pharmaceutical Inc. (USA) (currently inactive).27,28 These registry systems acquire and accumulate information on general characteristics, symptoms, and medical history of the disease to better understand GNE myopathy. The number of patients has been estimated to be approximately 400 in Japan29,30 and at least 40,000 worldwide. 30
Clinical presentation
In general, symptoms of GNE myopathy appear in those aged from 10 to 40 years.29,31 The initial site of presentation is typically the tibialis anterior with associated muscle weakness, and the initial symptoms are frequently foot drop, difficulty in lifting toes, and stumbling, but it should be noted that patients may complain of difficulty in walking. 25 Muscle weakness gradually progresses to proximal and upper extremity muscles. The most distinctive clinical feature is that the quadriceps femoris remains unaffected until the advanced stages of the disease, 31 which assists in the diagnosis of GNE myopathy. 3 However, there are atypical patients who present predominantly with proximal muscle impairment and asymmetric hand weakness.32,33 Eventually, patients become non-ambulatory and need a wheelchair an average of 10 to 20 years after disease onset.2,25,31
Histopathology
Histopathological features in the affected skeletal muscles in GNE myopathy include many rimmed vacuoles intensely stained with acid phosphatase and tubulofilamentous inclusions, and variation in muscle fiber size due to muscle atrophy.2,6,29,32 In most cases, there are no apparent findings indicative of muscle fiber necrosis.4,5 Inflammation is not usually found, although inflammatory cell infiltration has been reported in some cases.34–36
Extra-muscular symptoms
Blood tests
In patients with GNE myopathy, serum creatin kinase is at normal or mildly increased levels but tends to decrease with disease progression.2,3,37 Two siblings with myopathy with rimmed vacuoles and congenital thrombocytopenia who harbored two compound heterozygous GNE mutations, p.V603L and p.G739S, were reported. 38 Thrombocytopenia has occasionally (4.1% 39 ) been reported in patients with GNE myopathy.36–39 Among Japanese patients, the prevalence of thrombocytopenia was much higher than the estimated prevalence rate of idiopathic thrombocytopenia in the general Japanese population.39,40 Molecular mechanisms of decreased platelets may be explained by reduced sialylation activity in patients owing to the GNE gene mutations. Hyposialylation of surface materials on platelets may lead to shortened platelet life span.40,41 Recently, elevation of platelet-associated immunoglobulin G (PA-IgG) has been detected in the majority (65.4%) of examined GNE patients. The PA-IgG titer is expected to be a candidate as a surrogate biomarker for GNE myopathy (Section “Biomarker and natural history study”).
Respiratory dysfunction
Respiratory muscles are thought to be relatively unaffected in patients of GNE myopathy. However, one-third of patients had reduced respiratory function (percent forced vital capacity [%FVC] < 80), and two patients needed respiratory care with nocturnal non-invasive positive pressure ventilation. 42 A recent natural history study in Japanese patients demonstrated that %FVC was significantly decreased during a 5-year observation period. 37 Of note, decrease in %FVC in non-ambulatory patients was greater than that in ambulatory patients. 43 Although no correlation has been found between %FVC and disease duration, 39 it is clinically important to follow respiratory function, especially in non-ambulatory patients.
Sleep apnea syndrome
Mild to severe sleep apnea syndrome (SAS) was frequently observed in Japanese patients with GNE myopathy. 40 A nationwide questionnaire survey revealed that the prevalence of SAS was 10.4% in Japanese patients who were registered in REMUDY as having GNE myopathy. 39 In this survey, no correlation was identified between SAS and other patient characteristics including age, disease duration, body mass index, and %FVC. The 237th ENMC International Workshop in 2018 did not mention SAS in the context of GNE myopathy. 44 The clinical phenotype of GNE myopathy does not align with typical SAS risk factors such as age, high BMI, or waist circumference, suggesting that the disease itself may need to be recognized as a risk factor. It is recommended to monitor SAS in patients with GNE myopathy.
Cardiac involvement
Cardiac abnormalities have been detected by electrocardiography (ECG), echocardiography, and Holter ECG in some patients with GNE myopathy.40,43 So far, there have been no reports indicating the existence of a statistical association between these abnormalities and GNE myopathy. However, reports of sudden death in siblings with arrhythmia underscore the importance of yearly basic screening with ECG.40,43,45
Pregnancy
Regarding pregnancy and delivery in women with GNE myopathy, the incidence of threatened abortion is higher compared to that of the general Japanese population. 46 In addition, muscle weakness progressed after delivery in some cases, suggesting the clinical significance of perinatal management.
Diagnosis
Clinical manifestations, electrophysiology, histopathology, and muscle imaging are diagnostic of GNE myopathy.47–52 However, a definitive diagnosis is made by identification of GNE gene mutations. In clinical practice, it should be noted that a small portion of patients present with atypical clinical manifestations (Section “Clinical presentation”), thus leading to an increased risk of undiagnosed GNE myopathy.29,53 Moreover, a hallmark of GNE myopathy, relatively spared quadriceps, becomes evident in the later stages of the disease, which may hinder an early diagnosis. As the aim of developing therapeutic agents for GNE myopathy is to slow and ultimately prevent progressive muscle weakness (Sections “Clinical studies of aceneuramic acid replacement therapy” and “Ongoing clinical studies”), it is important to develop a more reliable and convenient diagnostic procedure for detecting the disease in its early stages.
Development of aceneuramic acid and replacement therapy
Fundamental studies
Etiology
Sialic acids are widely distributed in the natural world as components of glycoproteins and glycolipids. 2 Within living organisms, sialic acid-containing sugar chains on cell surfaces play important roles in various biological processes. 53 N-acetyl-neuraminic acid (aceneuramic acid, NeuAc) is a common form of sialic acid in human tissues. 2 In the sialic acid biosynthetic pathway, the GNE gene encodes a rate-limiting enzyme, uridine diphosphate (UDP)-N-acetylglucosamine (GlcNAc) 2-epimerase /N-acetylmannosamine (ManNAc) kinase. 54 This enzyme is composed of two domains having epimerase and kinase activities, respectively, and is essential for the in vivo sialylation of glycoproteins and glycolipids.
GNE myopathy is an autosomal recessive hereditary disorder caused by biallelic mutations in the GNE gene. There are homozygous or heterozygous missense mutations in the GNE gene, which diminishes overall sialylation activity of the GNE gene product.55–58 The functional missense mutations in the GNE gene lead to a sialic acid deficiency and reduced sialylation in body tissues, followed by protein aggregation and inclusion body formation, and presumably significant impairment of physiological functions.59–63 There are studies indicating that GNE mutations alter interaction with alpha-actinin2, 64 suggesting that functions beyond sialic acid synthesis await further investigation.
Some studies have indicated a correlation between genotype (mutation site) and phenotype (age at onset, severity, progressive rate). For example, Pogoryelova et al. reported that differences in GNE genotype account for 20% of variability in phenotype such as age at onset. 65 Mori-Yoshimura et al. suggested patients with homozygous p.V603L (previous p.V572L 16 ) mutations in the kinase domain had earlier onset and loss of ambulation compared to patients with compound heterozygous mutations in the epimerase/kinase domains. 66 However, whether a genotype-phenotype correlation exists is still an open question and remains to be investigated in future studies. Several amino acid substitutions by GNE missense mutations appear to be common in certain ethnicities or regions: Middle Eastern (p.M743 T),67,68 Japanese (p.V603L, p.D207 V, p.C44S),69–71 Roma Bulgarian (p.I618 T), 72 and Indian (p.V727 M). 53 Pathologic variants have been also identified in unusual regions of the GNE gene, including the region containing the 5’ untranslated region (5’UTR) in Japanese patients 73 and in the promoter region. 74 RNA sequence analysis has been useful for genetic characterization in some cases of GNE myopathy. 75
In vitro sialic acid supplementation to patient-derived muscle cells
Sialic acid levels in muscles and primary cultured cells derived from patients were significantly decreased compared to normal control cells. 55 Supplementation of NeuAc and ManNAc (a precursor of NeuAc) to the cultured cells resulted in an increase of sialic acid to normal levels, suggesting that the externally added NeuAc and ManNAc may compensate for the intrinsic deficiency of sialic acid in the muscles of patients with GNE myopathy.
Sialic acid supplementation to a mouse model of GNE myopathy
An early attempt to generate a GNE gene-knockout Gne(-/-) mouse was not successful, 76 suggesting that the GNE gene is essential for survival. Therefore, a transgenic mouse (hGNED176V-Tg) expressing the mutated human GNE gene that is common among Japanese patients (p.D207 V [previous p.D176 V]) 16 was first generated. The hGNED176V-Tg mouse was crossed with a Gne(+/-) mouse to yield a Gne(-/-)hGNED176V-Tg mouse.76,77 The resulting mouse exhibited reduced levels of sialic acid in serum and muscles, presenting with pathological features characteristic of GNE myopathy including muscle atrophy and weakness, thereby leading to its definition as a GNE myopathy mouse model.
The mouse model was used in a pre-clinical experiment of sialic acid supplementation. 78 Mice were treated with oral NeuAc or ManNAc from 10–20 weeks of age before occurrence of muscle weakness until 54–57 weeks of age. Administration of NeuAc or ManNAc led to an elevation in serum sialic acid concentrations and retention of muscle contractile force and motor performance equivalent to normal mice. Treatment with NeuAc or ManNAc also prevented pathological abnormalities including muscle atrophy and decrease in muscle fiber diameter. Rimmed vacuoles seen in muscle cells of control model mice were scarcely observed in the treated mice. These findings suggest that sialic acid supplementation may prevent the onset and/or progression of GNE myopathy in humans. As in the above mouse model, low levels of sialic acids in serum and tissues were reported in other mouse models of GNE myopathy. Toxicity, safety, and pharmacokinetics analyses of the NeuAc-containing formulation have been completed in pre-clinical studies using animals and microorganisms. Additionally, oral supplementation of peracetylated ManNAc and sialyllactose has been shown to improve skeletal muscle phenotypes in GNE myopathy model mice.79,80
Clinical studies of aceneuramic acid replacement therapy
Completed studies
Phase I studies in Japan and the USA
We started a first-in-human phase I study (NCT01236898) in 2010 in which aceneuramic acid was administrated to Japanese patients with GNE myopathy. 81 However, multiple doses (2400 mg/day) of aceneuramic acid for 5 days did not increase serum total aceneuramic acid concentration. Ultragenyx Pharmaceutical developed a new formulation, an extended-release aceneuramic acid (SA-ER) tablet. In 2011, Kakkis et al. conducted a phase I study (UX001-CL101; NCT01359319) 82 using SA-ER. Administration of SA-ER (maximum dose of 6000 mg/day) to patients resulted in an increase in serum free aceneuramic acid concentration, with no significant safety issues. Subsequently, we also conducted the second phase I study (UMIN000011532) in which SA-ER tablets (aceneuramic acid, 6000 mg/day) were administered to patients. Repeated dosing of SA-ER for 7 days achieved 2.4 times higher serum free aceneuramic acid concentration compared to pre-administration. All the adverse events (AEs) reported in the study were mild in severity and deemed unrelated with SA-ER, thus indicating the safety and tolerability of aceneuramic acid 6000 mg/day.
International phase II study
Argov et al. conducted a phase II, randomized, double-blind placebo-controlled study (UX001-CL201; NCT01517880) in 47 patients with GNE myopathy at 4 sites in 2 countries (USA and Israel) to assess the safety and efficacy of SA-ER. 83 For the first 24 weeks, patients were treated with SA-ER 6 g/day (6 g group), SA-ER 3 g/day (3 g group), and matching placebo. For the subsequent 24 weeks, patients in the 6 g group and the 3 g group continued with the same treatment (6/6 g group and 3/3 g group, respectively), whereas patients in the placebo group were given SA-ER 6 g/day (0/6 g group) and 3 g/day (0/3 g group).
At Week 24, the upper extremity composite (UEC) score in the 6 g group was significantly maintained compared to the placebo group. At Week 48, UEC score in the 6/6 + 0/6 g group (6/6 g group and 0/6 g group combined) was significantly improved compared to the 3/3 + 0/3 g group (3/3 g group and 0/3 g group combined). The difference in UEC score between the 6/6 + 0/6 g and 3/3 + 0/3 g groups was greater in the subgroup with a 6-min walk test (6MWT) ≥ 200 m compared to the overall population. The results suggested that administration of SA-ER suppressed progressive muscle weakness in patients. At Week 48, the lower extremity composite (LEC) score in the 6/6 + 0/6 g group was numerically higher but it was not statistically different from that in the 3/3 + 0/3 g group. Lack of significance may be attributable to greater impairment in lower extremity muscles compared to upper extremity muscles at baseline. Consistent with this presumption, there were no significant effects of SA-ER in the 6MWT and gait speed test. The GNE myopathy-functional activity scale (GNEM-FAS) is a measure of changes in daily living-associated physical functions in patients with GNE myopathy. At Week 48, GNEM-FAS scores (total, mobility, and upper extremity scores) in the 6/6 + 0/6 g group were significantly higher than those in 3/3 + 0/3 g group, suggesting that SA-ER also inhibited decline in muscle function.
Regarding safety, AEs were generally mild and moderate in severity, and non-serious. The most common AEs were procedural pain (72.3% of patients) caused by muscle biopsy. Diarrhea was also reported in 23.4% of patients. Overall, treatment with SA-ER was safe and well tolerated.
Phase II/III study in Japan
We conducted a phase II/III, randomized, multicenter, placebo-controlled, double-blind study (Sialic-Acid_3; UMIN000020683) in Japanese patients with GNE myopathy to evaluate the safety and efficacy of SA-ER (6 g/day) treatment. 84 A total of 20 patients were enrolled at 5 sites in Japan. Of those, 18 patients were able to walk 200 m or more in the 6MWT; one patient was not. One discontinued the study due to detection of pregnancy. Patients were randomly assigned in a ratio of 4:1 to the SA-ER group or the placebo group. They then received SA-ER (6 g/day) or matching placebo, respectively, for 48 weeks.
The primary endpoint was change in UEC score from baseline at Week 48. Statistical analysis revealed that a decrease in UEC score in the SA-ER group was significantly suppressed compared to the placebo group, suggesting that administration of SA-ER to patients may prevent progressive muscle weakness. Data of secondary endpoints including GNEM-FAS scores (total, upper extremity, mobility, and self-care scores) and LEC score supported the result for the primary endpoint. The Japanese version was developed through a validation process that included forward and backward translation.
Overall, AEs were noted in 87.5% and 75.0% of patients in the SA-ER and placebo groups, respectively. The most commonly affected system organ classes were Infections and infestations and Gastrointestinal disorders. Fetal death was experienced by one patient who became pregnant but was pregnancy test-negative at the start of administration. Except for this case, AEs were considered mild to moderate in severity. No significant safety concerns were associated with SA-ER treatment.
Extension study in Japan
An extension study (Sialic-Acid_4; UMIN000026354) was conducted in patients who completed the previous Sialic-Acid_3 to further evaluate the long-term safety and efficacy of SA-ER treatment (manuscript in preparation). Both patients in the SA-ER and placebo groups in Sialic-Acid_3 were treated with SA-ER (6 g/day) in Sialic-Acid_4 (termed SA-ER/SA-ER group and Placebo/SA-ER group, respectively) for 72 weeks. 85
Change in UEC score from the start of Sialic-Acid_3 to the final evaluation point in Sialic-Acid_4 (−3.48 kg at Week 120 or at early termination) was numerically smaller in the SA-ER/SA-ER group than that in the Placebo/SA-ER group (−8.10 kg), which indicated that long-term treatment with SA-ER did not completely arrest disease progression but exerted some beneficial effects in terms of retaining muscle strength and slowing the progression of GNE myopathy.
During Sialic-Acid_4, AEs were reported in 78.9% of patients, and were mild to moderate. AEs related to the study drug included gastrointestinal symptoms of feces soft and constipation, both mild in severity. Administration of SA-ER in the SA-ER/SA-ER group over 120 weeks posed no significant safety concerns, indicating that long-term treatment with SA-ER would be safe and tolerable.
International phase III study
Lochmüller et al. conducted a large-scale phase III, double-blind, placebo-controlled, randomized, international, multicenter study (UX001-CL301; NCT02377921) to evaluate the efficacy and safety of SA-ER treatment. 86 A total of 89 patients who were able to walk 200 m or more during the 6MWT at screening were enrolled at 13 sites in 7 countries. Patients were randomly assigned in a ratio of 1:1 to receive either SA-ER (6 g/day) or matching placebo for 48 weeks. At Week 48, change in UEC score from baseline (primary endpoint) as well as change in LEC score, muscle strength in knee extensors and the GNEM-FAS mobility domain score from baseline (key secondary endpoints) were not significantly different between the SA-ER and placebo groups. As such, Ultragenyx Pharmaceutical decided to terminate development of SA-ER for treatment of GNE myopathy.
UX001-CL201 and Sialic-Acid_3 indicated significant efficacy of SA-ER based on UEC score, whereas UX001-CL301 did not. The lack of efficacy in UX001-CL301 could be explained by the following factors in UX001-CL30165,84,86: (1) difference in clinical environments and larger variation in evaluating outcome measures due to participation of many countries and sites; (2) more genetically heterogeneous patient background; (3) higher proportion of mildly impaired patients (use of walking devices: 33% in UX001-CL201 vs. 18% in UX001-CL301), likely resulting in smaller changes in outcome measures. In fact, decrease in muscle strength in the placebo group was smaller than the pre-assumed value during the treatment period; (4) Lower serum aceneuramic acid concentration at Week 48 in UX001-CL301 than concentrations in UX001-CL201 and Sialic-Acid_3.
SA-ER was well tolerated. The most common AEs were mild to moderate gastrointestinal disorders. Serious AEs (SAEs) were reported in only two patients in the SA-ER group (grade 3 acute myocardial infarction and grade 2 acute gastritis) and one patient in the placebo group (abortion). All the SAEs resolved. Only acute gastritis was considered to be related to the study drug.
Phase III efficacy confirmation study in Japan
Based on UEC score, a treatment benefit of SA-ER was observed in the Sialic-Acid_3 study in Japan but not in UX001-CL301. Therefore, the regulatory authority in Japan required investigators in Sialic-Acid_3 to present further scientific evidence of the effectiveness of SA-ER in a newly designed study based on a scientific and rational explanation for the lack of benefit in UX001-CL301. After consultation with the regulatory authority, we conducted a phase III, randomized, placebo-controlled, double-blind, parallel-group, multicenter efficacy confirmation study (NPC-09–1, NCT04671472) of SA-ER. In NPC-09-1, the efficacy of SA-ER was evaluated in a patient population that met two additional criteria including a GNEM-FAS upper extremity domain score of 24 points or more and disease duration of 5–15 years. 87
A total of 14 patients who met the above two criteria were enrolled at 5 sites in Japan and were randomly assigned in a ratio of 5:2 to receive SA-ER (6 g/day) or placebo for 48 weeks. UEC score decreased over time in the placebo group, whereas the decrease was relatively suppressed in the SA-ER group, consistent with the results of Sialic-Acid_3. The GNEM-FAS upper extremity score declined in both the SA-ER and placebo groups, but more slowly in the SA-ER group.
In total, 55 AEs were observed in 9/10 patients (90%) in the SA-ER group and 17 AEs in 4/4 patients (100%) in the placebo group. Gastrointestinal disorder AEs were reported in 60% and 50% of patients in the SA-ER and placebo groups, respectively. AEs were mild to moderate in severity except for 2 SAEs including COVID-19 and papillary thyroid cancer. No AEs were considered related to the study drug.
Meta-analysis of SA-ER treatment benefits from clinical studies
We have selected papers on the oral treatment of sialic acid in GNE myopathy that are published on PubMed as of July 2024. In Figure 1, the treatment benefit of SA-ER based on change in UEC score in clinical studies thus far completed is shown as a forest plot of least squares (LS) mean difference in change in UEC and its 95% confidence limit between the SA-ER and placebo groups. In all of the studies, administration of SA-ER (6 g/day) numerically or significantly suppressed loss of upper extremity muscle strength compared to placebo during the treatment period of 24 or 48 weeks, without unacceptable adverse effects. Overall, the meta-analysis indicates that SA-ER is effective in slowing progressive muscle weakness in GNE myopathy.
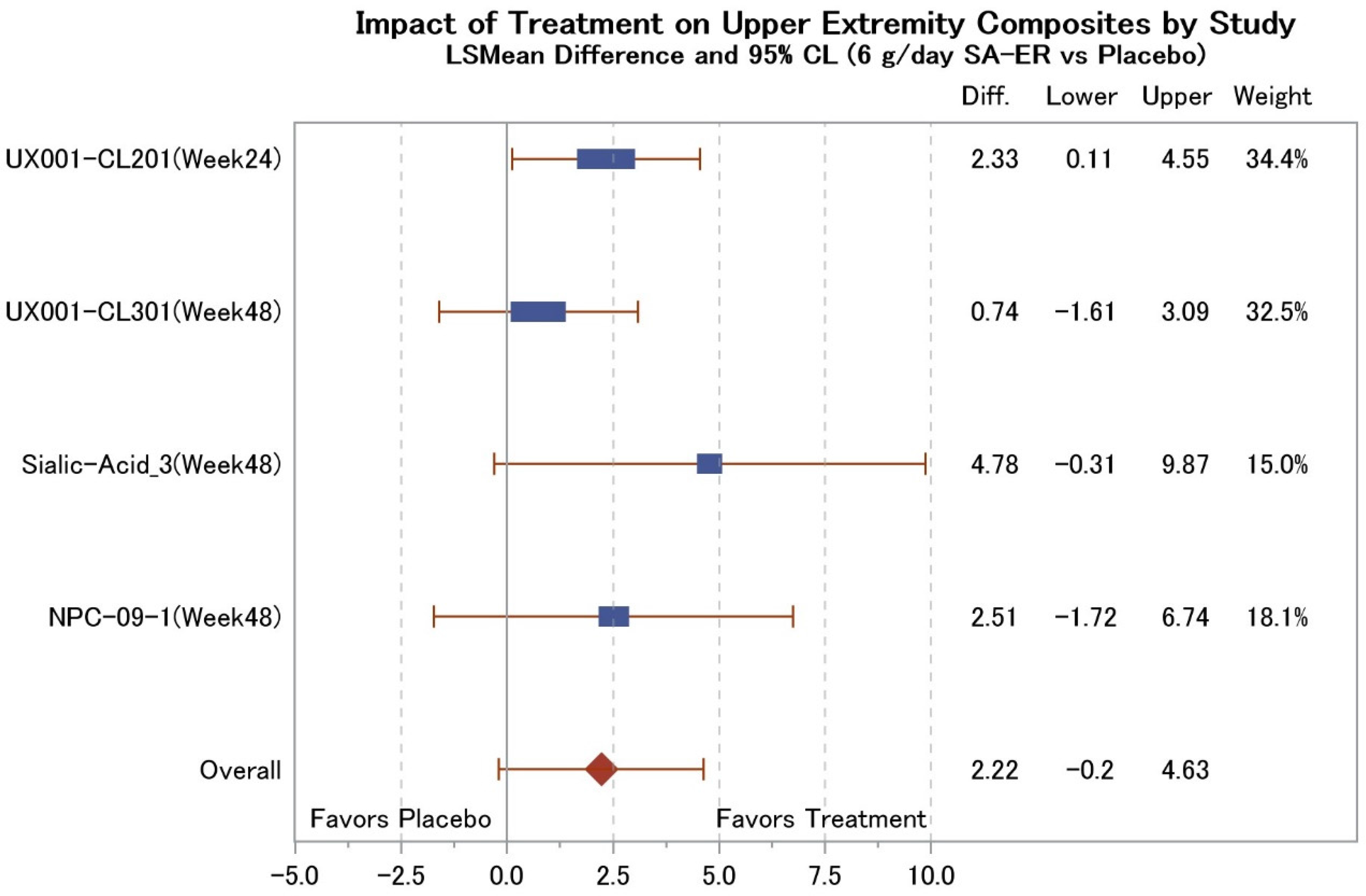
Impact of treatment on upper extremity composite by study. S mean difference (kg) and the corresponding 95% CL (6 g/day SA-ER vs. Placebo) are shown for each study using SA-ER and placebo. The overall effect of SA-ER was calculated by weighting with the reciprocal of the lower-upper range of 95% CL for group difference in each study. LS, least square; CL, confidence limit; SA-ER, extended-release aceneuramic acid.
Future perspectives
Ongoing clinical studies
There are 2 ongoing studies on GNE myopathy that have been registered in the ClinicalTrials.gov: one natural history study of GNE myopathy (NCT01417533) and one phase II study of ManNAc treatment (NCT04231266). ManNAc is expected to be another candidate for oral sialic acid supplementation. The previously conducted phase II 88 (NCT02346461) studies using ManNAc showed the safety and preliminary efficacy of ManNAc as compared to the natural history data on GNE myopathy. Currently, a phase II, randomized, double-blind, placebo-controlled, multicenter study (NCT04231266) is ongoing to evaluate the long-term safety and efficacy of ManNAc in patients with GNE myopathy. Pilot clinical trial to analyze the pharmacokinetic properties, safety, and efficacy of 6'-sialyllactose in patients with GNE myopathy was performed. 89 In the high-dose group, proximal limb powers improved with daily 6’-sialyllactose.
Biomarker and natural history study
GNE myopathy is a disease that progresses very slowly. The severity of impairment in muscles of the extremities differs considerably depending on the stage of the disease. Further, the rate of disease progression varies among patients. Therefore, it would be worthwhile to develop a biomarker that accurately indicates pathological changes and disease progression with high specificity and sensitivity, other than measures based on muscle strength and function. 1
Thrombocytopenia has been reported in some Japanese patients with GNE myopathy, with much higher frequency compared to the general Japanese population.1,39,40 Interestingly, PA-IgG was detected in 17 of 26 Japanese patients (65.4%) with GNE myopathy. Furthermore, there were significant differences in summed manual muscle testing (MMT), grip power, and platelet counts between PA-IgG-positive and PA-IgG-negative patients. 40 According to the findings, PA-IgG titer could serve as a potential surrogate biomarker reflecting the pathology of GNE myopathy. In addition, lower levels of Krebs von den Lungen-6 (KL-6) antigen in serum and tissues may be a useful indicator of reduced sialylation activity in patients with GNE myopathy. 90
The first prospective natural history study of GNE myopathy showed that summed MMT, grip power and %FVC were significantly reduced over a 1-year period. 43 A subsequent 3-year observational study reported that muscle strength (UEC and LEC scores) and function (GNEM-FAS scores) were significantly decreased from baseline during the study period. 91 In addition, the latter study indicated differences between Bulgarian and non-Bulgarian patients in muscle strength at baseline and decrease in muscle strength through Month 36, thus indicating the importance of considering the genetic background (GNE genotype) of patients in clinical studies. A recent 5-year natural history study also revealed a significant decrease in summed MMT, pinch power, gross motor function measure (GMFM) and %FVC over 5 years. Summed MMT can be performed easily compared to measuring the strength of multiple muscles with a hand-held dynamometer (HHD), thereby potentially generating more outcome data on the status of disease progression in routine medical practice and clinical studies. 37
Magnetic resonance imaging (MRI) evaluation of extremity muscle volume has been useful as a non-invasive method that reflects the pathological features of patients with GNE myopathy. 50 Recently, to accurately capture minute changes in muscles, Gidaro et al. assessed muscle images using quantitative nuclear MRI (NMRI) 92 (NCT02196909). Analysis of the cross-sectional area (CSA), contractile CSA (cCSA) and fat fraction (FF) at baseline revealed significant differences in CSA, cCSA and FF of lower extremity extensors between patients and healthy subjects. In addition, grip power correlated highly with cCSA and inversely with FF in flexor forearms. Follow-up assessment of muscle volume with quantitative NMRI successfully detected a significant change over a 1-year period. Interestingly, NMRI showed a significant decrease in cCSA with increase in FF in relatively spared quadriceps. Quantitative NMRI will allow physicians to assess pathological features more accurately and reproducibly in a rare muscle disease such as GNE myopathy, regardless of its disease progression status (i.e., ambulatory, or non-ambulatory).
Acquisition of real-world data
This section proposes what survey items might be recommended in post-marketing surveillance. The efficacy of aceneuramic acid supplementation in patients with GNE myopathy will be evaluated with real-world data based on the efficacy endpoints used in the phase III study in Japan (NPC-09-1) 87 : change in UEC score from baseline to Week 48, investigator-assessed efficacy rate, and changes from baseline to Week 48 in GNEM-FAS scores, each muscle strength of UEC composites (grip, shoulder abductors, elbow flexors, and elbow extensors) and knee extensors. However, HHD used for measuring muscle strength may be not available in every medical facility. As described in Section “Biomarker and natural history study,” summed MMT may be useful for efficacy evaluation of drug products.37,43 SAS frequently observed in Japanese GNE myopathy patients will be included in survey items.39,40 In addition, GNEM-FAS 87 and the 36-item short form survey (SF-36) 37 will be administered as part of post-marketing surveillance to assess the impact of muscle impairment on muscle function. As an objective measure complementary to UEC, muscle bulk will be assessed for flexor forearms by MRI imaging. 92 Treatment compliance will be verified with a patient diary. Pregnancy triggered worsening of conditions in some female patients. 46 This could be due to increased demand and depletion of sialic acid, as well as progression of muscle disuse atrophy due to bed rest. Careful administration is necessary, and it is important to assess whether exacerbation associated with pregnancy can be avoided with SA-ER.
Gene therapy
An alternative to oral sialic acid administration is gene therapy. 1 A recombinant adeno-associated virus (rAAV) harboring the human wild-type GNE cDNA was constructed to infect murine muscle cells and human primary muscle cells derived from patients with GNE myopathy. 93 The GNE mRNA was expressed in both cell cultures. The rAAV was injected into mice, resulting in successful expression of GNE mRNA in skeletal muscles for at least 6 months. In addition, single-dose administration of rAAV to a mouse model of GNE myopathy enhanced the expression of N-glycolylneuraminic acid (Neu5Gc) 94 in the murine muscles. For clinical development, assay methods were developed to determine the potency and dose of rAAV for GNE gene therapy. 95
Updates on clinical practice of GNE myopathy
As described in Section “Clinical studies of aceneuramic acid replacement therapy,” the three studies conducted in Japan (Sialic-Acid_3, Sialic-Acid_4, and NPC-09-1) consistently indicated the efficacy of SA-ER (identification code: NPC-09) treatment with no significant safety concerns in patients with GNE myopathy. In March 2024, the application for marketing NPC-09 (Acenobel) was approved by the regulatory authority in Japan. 96 The approval of NPC-09 has clinical significance as it is the first therapeutic medicine for GNE myopathy.
GNE myopathy patients routinely undergo conventional rehabilitation training and use assistive devices for maintenance and improvement of motor performance. In a recent move, Japan's health insurance system now covers the use of the robot suit HAL (Hybrid Assistive Limb, Cyberdyne Inc., Japan) 97 by GNE myopathy patients. In addition to such supportive devices, collaborative intervention by multiple professions is increasingly important for the comprehensive care of patients. Novel therapeutic agents will enable healthcare professionals to provide more focused care for patients.
Novel trends in drug development for orphan/ultra-orphan diseases
Not only in relation to studies on GNE myopathy, clinical studies on orphan/ultra-orphan diseases have the limitation of only a small number of patients being available. Moreover, it is ethically difficult to incorporate the concurrent placebo control group into the design of a clinical study particularly in the case of rare refractory diseases. To address such issues, a self-controlled study such as a crossover study and n-of-1 study can be planned. 98 If the natural history of a disease is well-known, an approach using historical data as an external control can be adopted.37,91,99 Recently, a Bayesian repeated measures model was developed to predict disease progression in GNE myopathy. 100
There are many additional challenges in the development of therapeutic agents for chronic progressive diseases requiring long-term observation. Particularly for an investigator-initiated study, it is necessary to create a more efficient process for securing sustainable sources of finance and an international coordination framework to catalyze the development of new therapeutic agents.
Recently, new attempts to submit an Investigational New Drug Application for a rare disease, amyotrophic lateral sclerosis (ALS), were made in the United States. One case involved the “compassionate use” pathway 101 of Food and Drug Administration (FDA) for unapproved drugs. Through this pathway, a therapeutic antisense oligonucleotide (ASO) was administered to a patient with FUS gene-mutated ALS.102,103 Another case involved the accelerated approval of an ASO drug Tofersen for treatment of SOD1 gene-mutated ALS. The phase III study of Tofersen (NCT02623699) failed to demonstrate efficacy in the primary endpoint.103,104 However, it was expeditiously approved by FDA based on an assessment made using a surrogate biomarker (neurofilament) for the disease. 105 In the case of a new safety-assured drug for a rare disease with a well-known natural history, it may be possible to obtain approval for use based on data for a surrogate biomarker and subsequently confirm the efficacy with real-world data acquired in clinical practice.
Taken together, it may be worth considering novel approaches for clinical studies and approval of therapeutic agents in the case of rare diseases after consultation with the regulatory authority in each country. Implementing these approaches could streamline therapeutic advancements, providing early access to novel treatments for patients.
Conclusion
Based on the favorable results of human clinical trials, an extended-release aceneuramic acid formulation was approved for treatment of GNE myopathy in Japan in March 2024. It is anticipated that it will be a significant step in the development of an effective treatment for GNE myopathy and other ultra-orphan diseases. As we move into an era where appropriate genetic diagnosis and therapeutic interventions are desired, it will be necessary to collect real-world data on long-term drug treatment.
Footnotes
Acknowledgements
We would like to extend our gratitude to all the patients and their families who participated in clinical trials, the Registry of Muscular Dystrophy (REMUDY), the Patients Association for Distal Myopathies (PADM), and researchers/clinicians involved in the development of the drug. We thank the Clinical Research Innovation and Education Center Tohoku University (CRIETO) for their help in scheduling during the study. Medical writing of this paper was supported by Tetsuyoshi Inoue, PhD and Mizuko Osaka, PhD (SunFlare Co., Ltd, Tokyo, Japan) and the support was funded by Nobelpharma Inc., Ltd
Contributors
NS drafted the manuscript. All authors revised the manuscript and gave final approval for publication.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Grants-in-Aid for Research on Rare and Intractable Diseases (23FC1014) from the Ministry of Health, Labour, and Welfare of Japan.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Ichizo Nishino is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review.