
Abstract
Background:
Myofibrillar myopathy (MFM) is a heterogeneous group of neuromuscular disorders characterized by degeneration of Z-disk and disintegration of myofibrils. OBJECTIVE: We aimed to analyze the mutational spectrum and phenotypic features of MFM in China.
Methods:
We used targeted next generation sequencing (NGS) to identify causative mutations in 39 MFM patients with confirmed myopathological diagnosis.
Results:
The results showed that variants were found in six MFM-associated genes, including DES, FLNC, BAG3, MYOT, TTN and DNAJB6, in 28 (71.7%), 3 (7.7%), 3 (7.7%), 1 (2.6%), 3 (7.7%), and 1 (2.6%), respectively. Of the total 26 variants identified, 19 were reported previously and 7 were novel variants. Missense variant (80.0%) was the most common mutant type of DES. P209L was the hotspot mutation of BAG3 while no obvious hotspot mutation was found of DES. Clinically, distal and proximal weakness were observed in 64.1% and 35.9% patients. Arrythmia and peripheral neuropathy were the most common combined symptoms of desminopathy and BAG3opathy, respectively. Pathologically, rimmed vacuoles (RVs) were present in different genetic type of MFM. Giant axonal nerve fiber was found in BAG3-releated MFM patient.
Conlusion:
We concluded that MFM showed a highly variable genetic spectrum, with DES as the most frequent causative gene followed by FLNC, BAG3 and TTN. This study expanded the genotypic and phenotypic spectrum of MFM among Chinese cohort.
Introduction
Myofibrillar myopathy (MFM) is a clinically and genetically heterogeneous group of hereditary myopathies characterized by myofibril dissolution and abnormal accumulation of degradation products. 1 The disintegration of the myofibrils commences in the immediate proximity of the Z-disc and results in Z-disc streaming in ultrastructural findings. 2 The abnormally accumulated proteins include desmin, myotilin, α-B-crystallin, filamin C, Bag3, actin, plectin, dystrophin, sarcoglycans, neural cell adhesion molecule (NCAM), gelsolin, ubiquitin, syncoilin, synemin, Xin, and TAR DNA-binding protein 43 (TDP-43). Presence of rimmed vacuoles (RVs) associated with degraded aggregates are observed on histological staining with hematoxylin and eosin (HE) and modified Gomori trichrome (MGT). 3
Despite the common histological features, the clinical manifestations of various subtypes can change and may include different age of onset (from childhood to late adulthood), distal more than proximal weakness (only 25% patients with early limb-girdle involvement), cardiopathy, respiratory failure, cataracts or peripheral neuropathy in various combinations. 4 Due to such significant phenotypic variability, the clinical diagnosis of MFM is often difficult.
In recent years, the application of genetic technology provided a great deal of advantages for screening multiple candidate genes parallelly. MFM patients with genetic diagnosis have been reported worldwide. MFM is usually inherited in an autosomal dominant manner; however, autosomal recessive or X-linked MFM forms have also been described. 5 The majority of genetically diagnosed patients harbor pathogenic mutations in one of six genes: desmin (DES), α-B-crystallin (CRYAB), myotilin (MYOT), LIM domain binding 3/Z band alternatively spliced PDZ-containing protein (LDB3/ZASP), filamin C (FLNC), and Bcl2-associated athanogene-3 (BAG3). In addition, mutations in genes typically associated with other muscular diseases can also result in MFM-like clinical or pathological phenotypes. These genes were considered as MFM-related genes according to 2022 version of the gene table of neuromuscular disorders and previous reports,6,7 including TTN, KY, PYROXD1, FHL1, DNAJB6, PLEC, ACTA1, HSPB8, LMNA, SQSTM1, SVIL and UNC-45B and are still expanding (Table 1).8–19 Each gene produces a mutated protein which is an integral part of the Z-disk or is closely associated with it. 1
Genes related to MFM-like phenotype.

AD, Autosomal dominant; AR, Autosomal recessive.
Herein, we subjected a large cohort of Chinese MFM patients to targeted next generation sequencing (NGS), to investigate their mutational spectra. In addition, their genotype–phenotype correlation was also analyzed.
Materials and methods
Patients
Muscle biopsy results of 10,509 patients were reviewed at the Department of Neurology, Peking University First Hospital from 2001 to 2022. According to pathological findings and clinical features, 78 suspected MFM patients were screened and 49 of the 78 patients received genetic test. We retrospectively analyzed the NGS results. Finally, 39 (including individuals from 30 unrelated families and four pairs of siblings) of 49 patients (79.6%) were enrolled with confirmed MFM-related genes variants (Figure 1). Four of them had been reported by us previously (F1 and F2). 20 Their enrollment was based on the following inclusion criteria: (1) slowly progressive muscle weakness or cardiac symptoms; (2) eosinophilic deposits on HE staining, RVs on MGT staining and positive sarcoplasmic immunostaining for MFM-related proteins; (3) confirmed MFM-related genes mutation. The clinical data were retrospectively collected from the patient medical records.
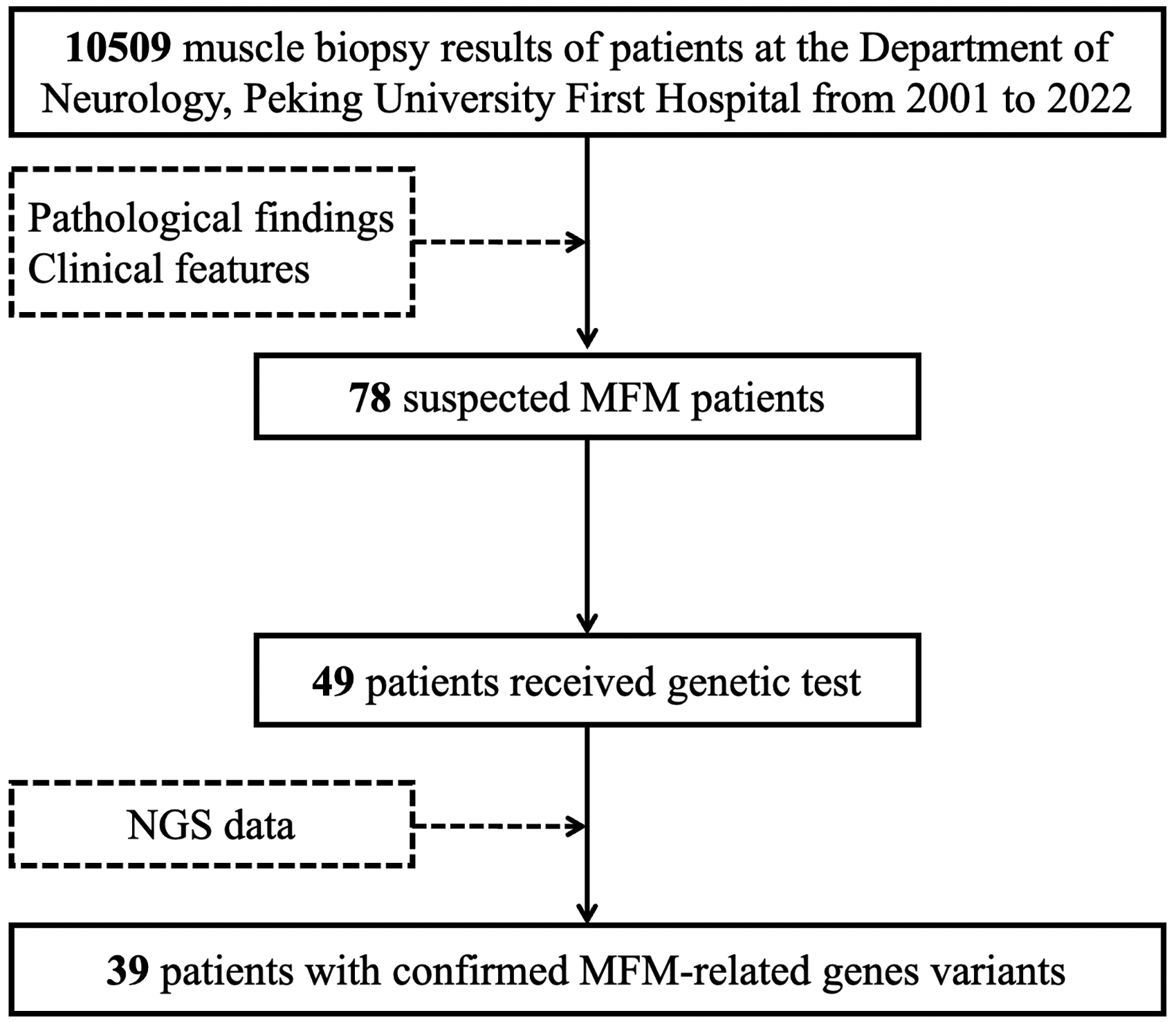
Flowchart of enrollment procedure.
All patients provided their informed consent for this study, and ethical approval was obtained from the Human Research Ethics Committee, of Peking University First Hospital.
Pathological analysis
The muscle biopsies were evaluated by two independent evaluators (YY and WZ), both of whom were experienced in interpreting muscle biopsies and muscle immunoanalysis results, and were blinded to genotypes of the patients. The muscle specimens were frozen in isopentane, cooled in liquid nitrogen, and then stored at - 80°C. 21 Routine histological and histochemical staining, using techniques involving HE, mGT, nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR), and adenosine triphosphatase at varying pHs, were performed, and standard techniques were used for immunohistochemical staining including antibodies against desmin (Abcam, 1:400). 21
For the analysis of the proportion of fibers with aggregates or RVs, 800–1000 fibers of muscle sections were counted in each patient. The percentage of fibers containing RVs was calculated, and divided into four grades: few (<1%), several (1%–5%), some (6%–25%) and many (>25%).
Molecular analysis
Genomic DNA was extracted from peripheral blood samples or muscle tissues taken from all patients using standard procedures. Sequence variants were detected using an NGS diagnostic panel with a minimum coverage of 100× to analyze all exons and their flanking sequences of genes associated with myopathies (Table S1). Sanger sequencing was performed using specific primers to confirm the variants detected by NGS.
Segregation analysis of the variants was performed in the available family members of 15 patients. Upon assessing the frequencies of variants in large populations, 100 healthy control participants (100HC) of Chinese origin were screened, and we also checked for allele frequencies in the Genome Aggregation Database (gnomAD), NHLBI Exome Sequencing Project (ESP6500) Exome Variant Server, 1000 Genomes Project (TGP), and Exome Aggregation Consortium (ExAC). The evidence of pathogenicity was deemed to be moderate (PM2) for variants that were absent or present at extremely low frequencies, at an alternative allele frequency of <0.5% in population databases. Multiple pieces of computational evidence were derived from various in silico analyses, where FATHMM, Mutation Taster, PolyPhen-2, and SIFTwere used to predict deleteriousness, GERP was used to assess evolutionary conservation, and GenomeBrowse was used to predict REVEL value. The impact of splicing in a variant spanning the exon and intron region was determined by the Human Splicing Finder (HSF). We used the wInterVar tool to automatically generate predictions regarding 6 (PS1, PM1, PM5, PP2, BP1, BP7) of 28 criteria specified in the ACMG-AMP guidelines; SVI General Recommendations for Using ACMG/AMP Criteria was used to improve consistency in usage and transparency in classification rationale; the rest were interpreted by manual review and adjustment, on the basis of detailed information about variants (such as the de novo status of a variant) and our own domain knowledge. These criteria were then combined, to arrive at a final interpretation.
Results
Clinical data
A total of 39 MFM patients from 34 families, including 22 males and 17 females, were enrolled in our study; their diagnosis was confirmed by myopathological and genetic findings. The onset age was 28.5 ± 16.1 years (range 1–59 years old; five DES mutant patients manifested with feeding difficulties and delayed motor milestones after birth were defined as 1 year old onset). Twenty-one patients reported a family history suggestive of neuromuscular disorders and among these four pairs of siblings were present (Table 2).
The clinical features of patients with MFM.
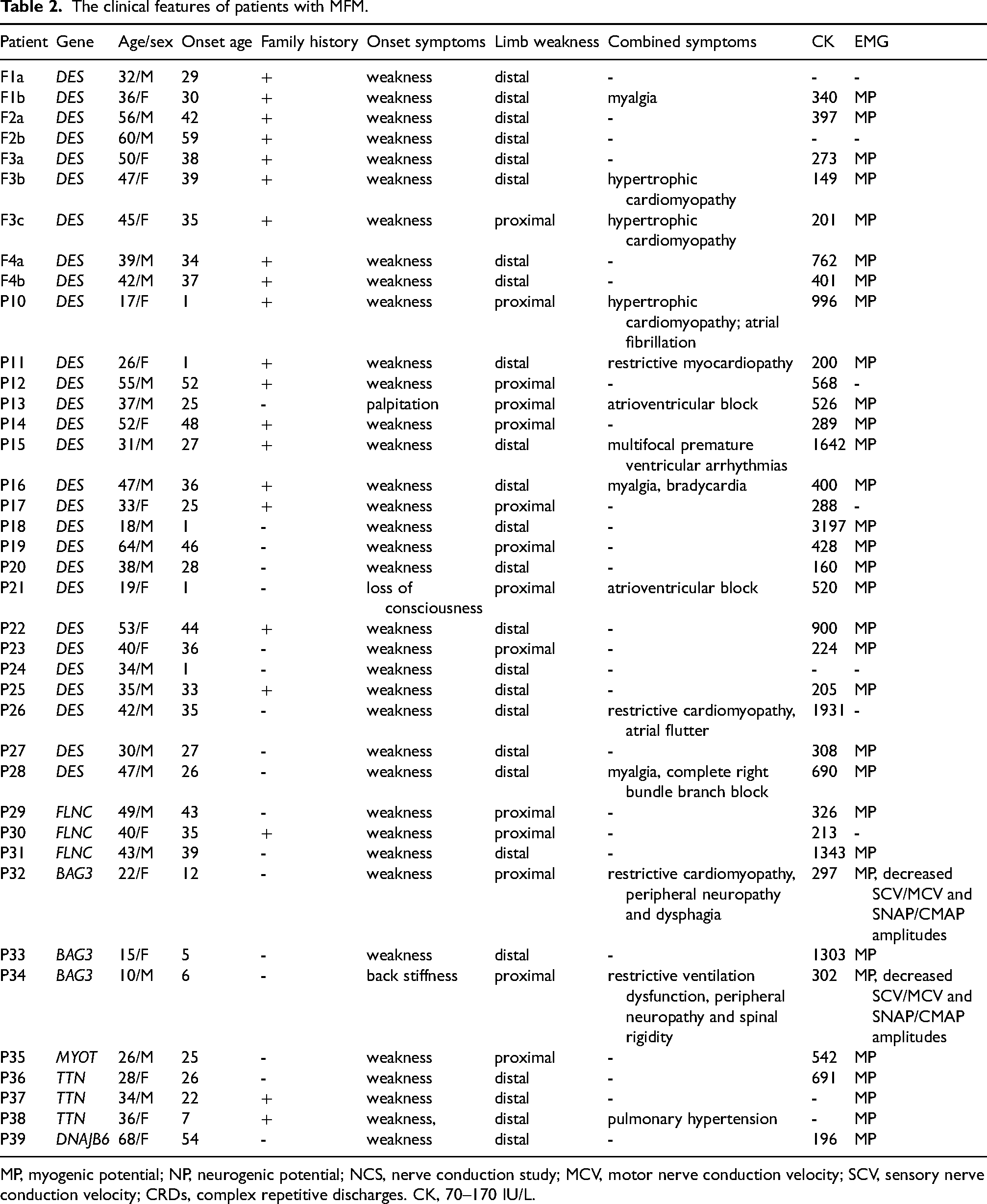
MP, myogenic potential; NP, neurogenic potential; NCS, nerve conduction study; MCV, motor nerve conduction velocity; SCV, sensory nerve conduction velocity; CRDs, complex repetitive discharges. CK, 70–170 IU/L.
The onset manifestations were weakness in 36 patients, cardiac symptoms in two DES mutant patients and back stiffness in one BAG3 mutant patient. Five DES mutant patients with onset age of 1 year old manifested with weak sucking and dysphagia after birth and delayed motor milestones. The age at onset of gait was 30, 29, 30, 24 and 28 months of P10, P11, P18, P21 and P24, respectively. P10 and P11 also presented with intermittent cyanosis when feeding and crying in infancy. Limb weakness was observed in all patients, and distal weakness (64.1%) was more common than proximal weakness (35.9%). Other combined symptoms found at the time of muscle biopsy in DES mutant patients consisted of arrythmia, myocardiopathy and myalgia (7, 5 and 3 patients, respectively). All three patients harbored FLNC mutations only presented with muscle weakness. Two of three patients with BAG3 mutations manifested with peripheral neuropathy, one with cardiopathy and dysphagia, and the remaining one with spine rigidity and respiratory dysfunction. Muscle weakness appeared in all patients with MYOT, TTN, and DNAJB6 variants. Notably, P39 carried TTN variant combined with pulmonary hypertension. Serum creatine kinase (CK) was mild elevated. All patients underwent electromyography showed myogenic changes. Two of three BAG3 mutant patients revealed abnormal nerve conduction study, presented with decreased SCV/MCV and SNAP/CMAP amplitudes.
Genetic results
Thirty unrelated patients and four pairs of siblings were identified to carry variants in one of 6 MFM-related genes including DES(n = 28),FLNC(n = 3), BAG3 (n = 3), MYOT(n = 1), TTN(n = 3) and DNAJB6 (n = 1) (Figure 2). In total, 26 variants were identified in these 6 genes, of which 19 were reported and 7 were novel ones (Table 3). The clinical interpretations of novel candidate variants detected in DES, FLNC, BAG3, MYOT, TTN, and DNAJB6 according to the American College of Medical Genetics and Genomics (ACMG) criteria has been summarized in Table 3.
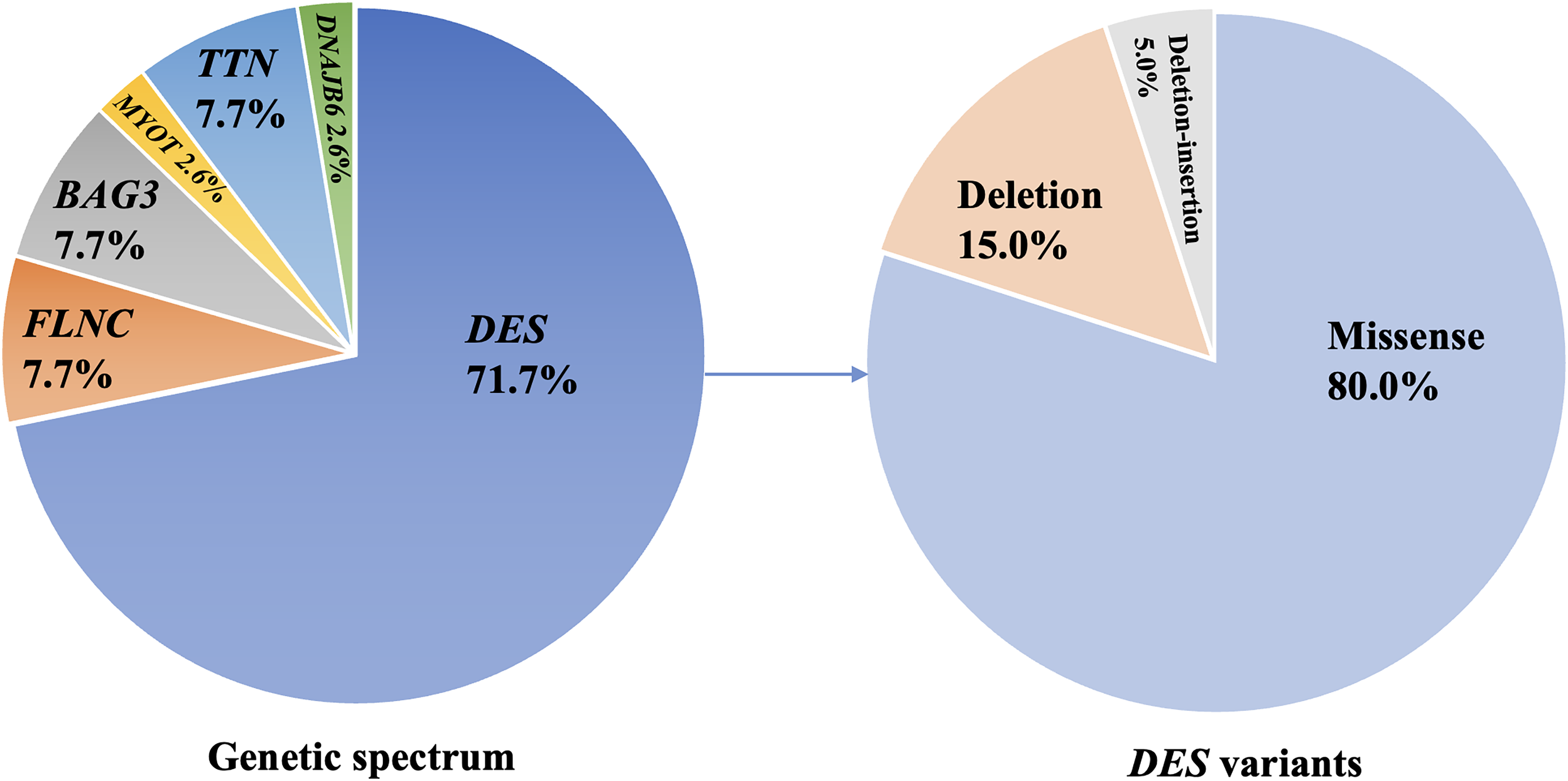
Genotypic spectrum of 39 MFM patients and mutation types of DES.
The genetic data of patients with MFM.
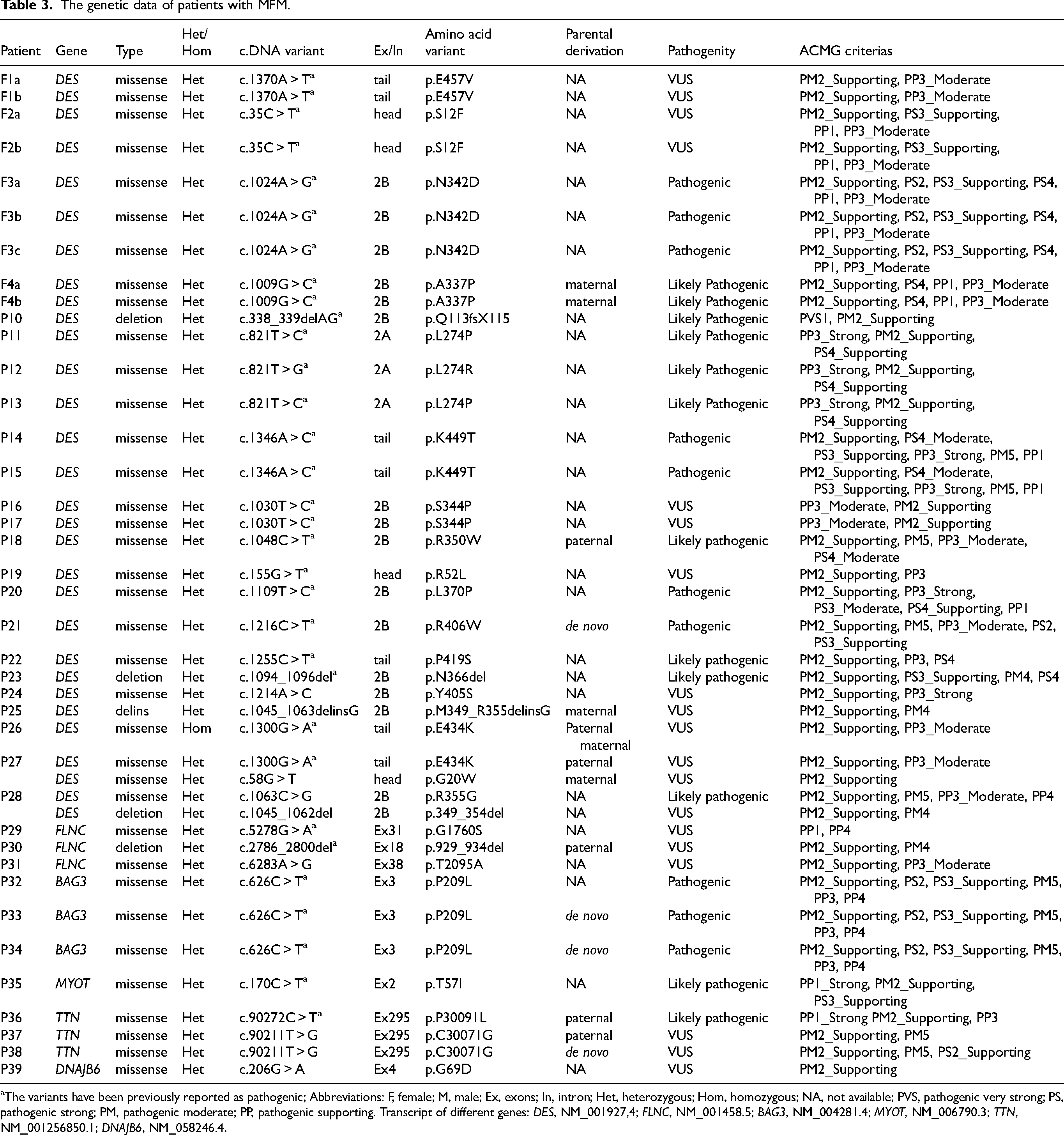
The variants have been previously reported as pathogenic; Abbreviations: F, female; M, male; Ex, exons; In, intron; Het, heterozygous; Hom, homozygous; NA, not available; PVS, pathogenic very strong; PS, pathogenic strong; PM, pathogenic moderate; PP, pathogenic supporting. Transcript of different genes: DES, NM_001927,4; FLNC, NM_001458.5; BAG3, NM_004281.4; MYOT, NM_006790.3; TTN, NM_001256850.1; DNAJB6, NM_058246.4.
DES
Twenty variants were identified in DES in 19 individuals and four pairs of siblings, including 16 missense variants, three in-frame variants caused by two deletions and one deletion-insertion, and one frameshift mutation caused by deletion (Figure 1). Except for P26 carrying a homozygous missense mutation, P27 carrying two compound heterozygous missense variants (one missense mutation c.1300G > A/p. E434K was same as the homozygous mutation in P26), P28 carrying one missense and one deletion variants without segregation analysis, other patients harbored single heterozygous variants of DES.
Among the variants, the three missense variants (c.1214A > C/p.Y405S, c.58G > T/p.G20 W and c.1063C > G/p.R355G), the deletion variant (c.1045_1062del/p.349_354del) and the deletion-insertion variant (c.1045_1063delinsG/p.M349_R355delinsG) were novel variants, the remaining 15 mutations have been reported previouly. 22 c.1063C > G/p.R355G were classified as likely pathogenic variant. The pathogenicity of the remaining novel variants was of undetermined significance (VUS). The most frequent mutation was c.821T > C/p. L274P carried by three individuals, no obvious hotspot mutation was found in DES.
For the distribution of the variants, three of them located at head domain, one at 2A segment, twelve at 2B segment and four at tail domain.
FLNC
We identified three variants in FLNC in three patients, including two missense (c.5278G > A/p.G1760S and c.6283A > G/p.T2095A) and one deletion (c.2786_2800del/p.929_934del) variant. The missense (p.G1760S) and the deletion mutation were reported previously, 23 the remaining (p.T2095A) was novel variant was of undetermined significance. The deletion mutation in P30 was parental inheritance, the others were single heterozygous without familial analysis.
BAG3
In accordance with most other BAG3opathy cases, 24 the missense mutations c.626C > T/p.P209L in exon 3 of BAG3 were identified in three BAG3opathy patients, of which two were de novo ones confirmed by segregation analysis.
MYOT
NGS revealed one heterozygous missense mutation c.170C > T/ p.T57I in one patient, and was reported as pathogenic mutation previously. 25
MFM-related genes
We also found variants in TTN and DNAJB6 in patients with MFM-like clinical and pathological features. DNA sequencing and family co-segregation analysis proved two heterozygous missense mutations (c.90272C > T/p.P30091L and c.90211T > G/ p.C30071G) in the same exon of TTN in three patients. The p.P30091L identified in P36 was a previously reported mutation. 8 The novel missense variants 206G > A/ p.G69D of DNAJB6 harbored by P39 were of undetermined significance.
Pathological findings
Table 4 documented myopathic features in different genotypic groups. Different percentage of fibers harbored amorphous material that was eosinophilic on HE staining and dark blue on MGT, some accompanied by cytoplasmic bodies (Figure 3). The percentage of muscle fibers containing deposits ranged from 1% to 70%, and were classified into the few, several, some and many in 8 (8.3%), 6 (6.2%), 11 (31.3%), and 3 (1.3%) of DES-mutant patients, respectively. The major proportion of fibers containing RVs in different genes was less than 5%. Muscle fibers with disorganized myofibrillar network on NADH-TR staining were common in patients of different genotype (Figure 3). Other non-specific myopathic changes included slight fiber size variation and splitting fibers. The lipid and glycogen content of muscle fibers were normal. In 76.5% muscle biopsies from patients with DES mutation and 67.5% of other genes, abnormal fibers with focal areas of increased aggregation of desmin were observed (Figure 3). The percentage of abnormal fibers was variable. In addition to the myopathic changes, patients with BAG3 variants also presented with neurogenic changes on muscle pathology and giant axon nerve fiber on sural nerve biopsy of P33 (Figure 3).
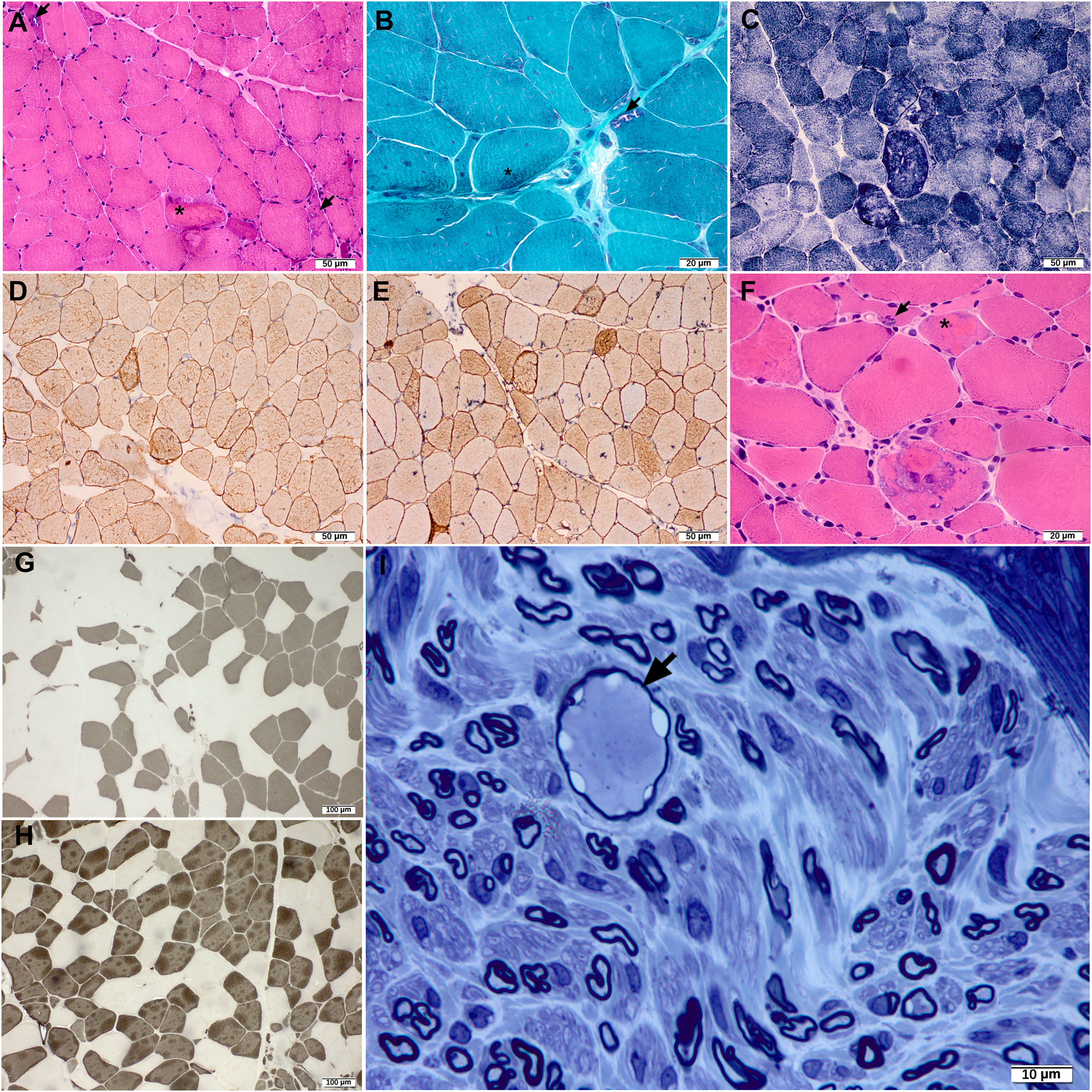
Myopathological features of P21 with DES (A-E), P30 with FLNC (F) and P32 with BAG3 (G-I) variants. (A and F) HE and (B) MGT staining showed amorphous material (asterisk) and RVs (arrow) in myofibers. (C) NADH staining revealed rubbed-out fibers. (D) Desmin and (E) dysferlin staining showed increased reactivity in abnormal myofibers. (G and H) ATPase staining revealed groups of atrophic type 1 and type 2 fibers. (I) Sural nerve biopsy of P32 showed giant axon (arrow).
Summary of muscle biopsy findings.

The MFM-like pathological findings in P24 (DES, p.Y405S), P25 (DES, p.M349_R355delinsG), P28 (DES, p.R355G/p.349_354del), P31 (FLNC, p.T2095A), P38 (TTN, p.C30071G) and P39 (DNAJB6, p.G69D) with novel variants were presented in Figure 4 to support the pathogenicity. P37 and P38 with TTN novel variant showed necklace cytoplasmic bodies in myofibers consistent with TTN mutant myopathological features. 26
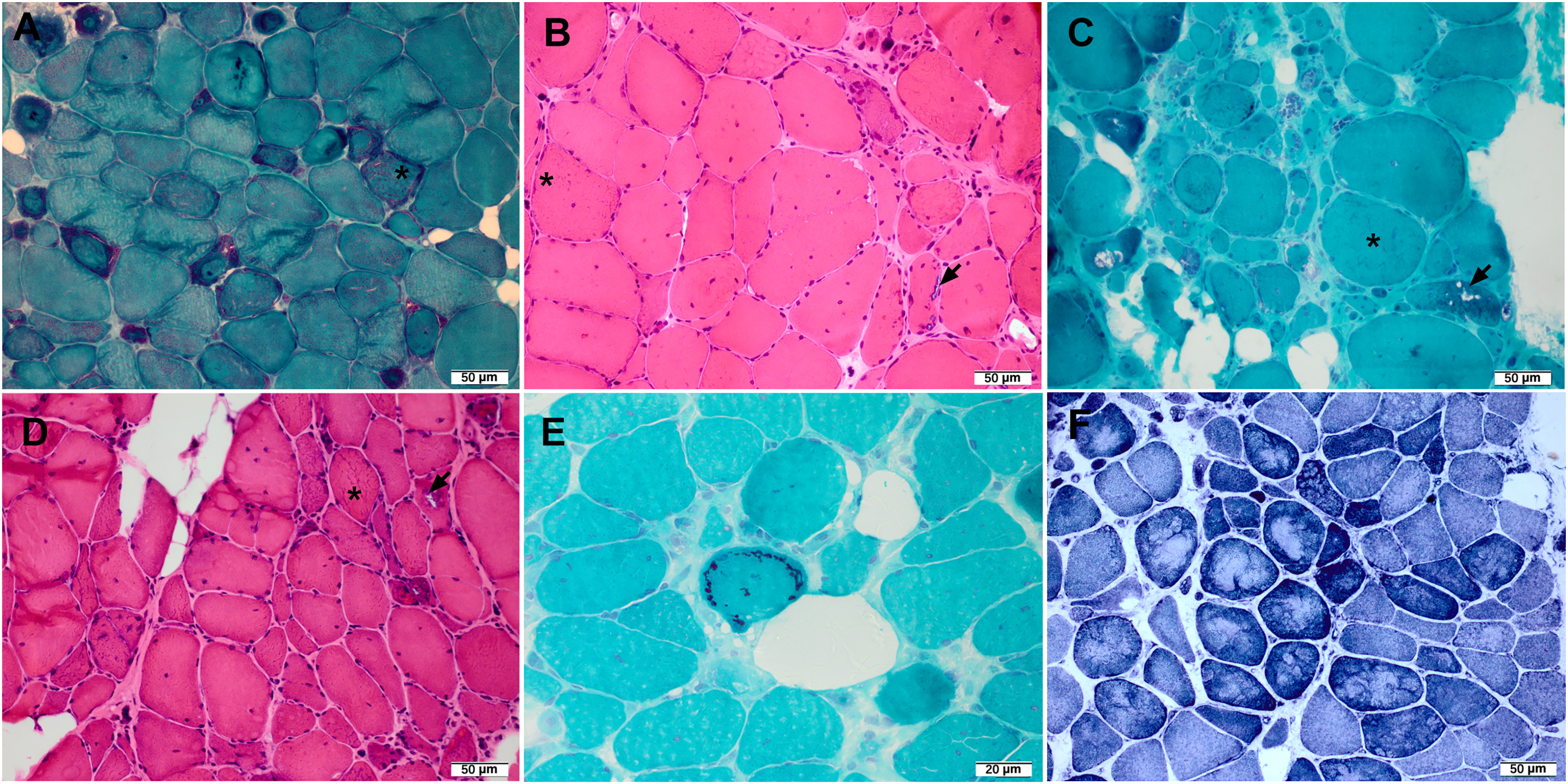
Myopathological features of patients with novel variants. (A) MGT staining of P24 with DES Y405S, (B) HE staining of P25 with DES M349_R355delinsG, (C) MGT staining of P28 with DES R355G/ 349_354del, (D) HE staining of P31 with FLNC T2095A showed amorphous material (asterisk) and RVs (arrow) in myofibers. (E) MGT staining of P38 with TTN C30071G showed necklace cytoplasmic bodies in myofibers, (F) NADH-TR staining of P39 with DNAJB6 G69D showed obvious oxidative enzyme abnormalities.
Discussion
We used targeted NGS to evaluate the mutational spectrum and prevalence of MFM in the Chinese population in a medical neuromuscular center in China. The enrollment of suspected MFM patients was based on clinical manifestations, myopathological changes and MFM-related gene mutations. In total, we identified causative mutations of six genes in 39 of 48 patients (79.6%), which was higher than the value of 50% in general. 7 Among the genes we screened, DES was the most common causative mutant gene, followed by BAG3, FLNC and TTN, which was compatible with another Chinese study. 27 Although the incidence and prevalence of desminopathies are currently not available, DES are the best-studied and most common entity of MFM. 28
DES encompassed 9 exons and was organized into 3 domains: the head (N-terminal structure), the α-helical rod (contained 1A, 1B, 2A and 2B segment), and the tail (C-terminal structure).29,30 Pathogenic variants were reported in all domains, but most were missense and located in segment 2B, small in-frame deletions and exon-skipping mutations have also been reported.2,31 The p.P419S substitution was reported as a high frequency mutation in Chinese cohort, appearing only in P22 in our cohort. 27 No obvious hotspot DES mutation was found in our study, in which the most frequent mutation was p.L274P identified in three unrelated patients. Deletion-insertion mutation haven’t been reported in DES. P25 with the novel c.1045_1063delinsG mutation shared the typical pathological features and clinical manifestations of MFM, broadening the genetic spectrum of DES. Familial primary desminopathies are usually autosomal dominantly inherited. Rare autosomal recessive DES variants are associated with an earlier clinical presentation (from childhood to early adulthood) and faster progression compared with more common autosomal dominant variants. 32 Andreas. B et al. reported the homozygous missense DES mutation p.Y122H (c.364T > C) in a patient with severe restrictive cardiomyopathy, 33 while homozygous nonsense mutation p. G234X and p.R150X was found to cause cardiac fibrosis and dilated cardiomyopathy, respiratory insufficiency, respectively,32,34 often without muscular complaints. Adult-onset P26 carried homozygous missense p.E434K, presenting with weakness, myocardiopathy and arrythmia. Compound heterozygous p.E434K was also identified in P27 combined with p.G20W, in which the muscle weakness was the predominant complaint. The missense mutation E434K was reported in a Chinese family presented with severe cardiopathy and arrythmia and inherited in autosomal dominant manner, 34 while E434K in P26 and P27 in our study was inherited in autosomal recessive manner and presented with earlier onset, slower progression of cardiac or muscular symptoms. We found E434K show significant phenotypic heterogeneity (spanning from cardiac defects to muscular weakness) across dominant and autosomal recessive subtypes, might be due to different penetrance, clinical variability, different genetic mechanisms or multiple modifier loci. The novel heterozygous p.349_354del and p.R355G variants in the same chromosome of P28 were defined as likely pathogenic mutations. The pathogenicity needed further investigation.
Mutated FLNC caused cardiomyopathy and/or myopathy either as myofibrillar myopathy or distal myopathy. 35 Consistent with previous study, most of pathogenic variants of FLNC responsible for myopathy are widely distributed in the actin-binding domain and Ig-like rod repeats. 36 c.5278G > A/ p.G1760S was reported to be related with proximal muscle weakness and dystrophic changes with RVs on biopsy.23,37 P29 with this mutation in our cohort presented with amorphous material and RVs in many fivers and ultrastructural Z-band streaming, diagnosed as myofibrillar myopathy, expanding the phenotype of this missense mutation. The deletion mutation p. 929_934del found in FLNC was reported to cause limb-girdle muscular dystrophy, 38 whereas in our case causing the clinical phenotypes of FLNC–related MFM.
The autosomal dominant variant P209L occurred at a high frequency of BAG3opathy. Consistent with other studies, all of the three patients carried missense c.626C > T/ p.P209L of BAG3, manifested with weakness, peripheral neuropathy, cardiomyopathy, respiratory failure and spine rigidity.39–41 c.626C > T/ p.P209L was previously reported to inherit with missense variant c.772C > T, while the carrier of c.772C > T was asymptomatic. 42 Neurogenic changes in muscle biopsy and myelinated nerve fibers with giant axon in nerve biopsy were pathological features of BAG3opathy.43,44
Myotilin binded to the Z-disc and filamin C and controlled sarcomere assembly. 45 Mutations in myotilin could cause MFM and exon 2 of MYOT was a hotspot for mutations. 46 p.T57I in exon2 was reported to be related to autosomal dominant form of limb girdle muscular dystrophy. 25 P36 with this mutation presented with MFM phenotype.
The inheritance pattern of TTN mutations can be autosomal dominant or recessive, or even the combination of both.47–49 TTN-related muscle pathology consisted of increased central nuclei and cores/minicores, which is more indicative of congenital myopathy than MFM.50,51 Heterozygous p.P30091L was reported in a Chinese family with hereditary myopathy with early respiratory failure, indicating the mutational vulnerability of TTN exon 295. 51 p.P30091L in P37 presenting with weakness but without respiratory dysfunction also inherited in an autosomal dominant manner. The novel missense mutation p.C30071G also located in exon 295 appeared in two of three TTN-mutant patients with MFM-like phenotype accompanied by necklace cytoplasmic bodies in myofibers, of which P39 accompanied by pulmonary hypertension not been reported in titinopathy. Whether the mutation and/or exon 295 was vulnerable to MFM needed further investigation.
Our cases highlighted the occurrence of MFM-like pathology in patient with DNAJB6 myopathy. DNAJB6 regulates chaperones by stimulating their ATPase activity. 48 DNAJB6 mutations usually caused limb-girdle muscular dystrophy type 1D. 52 DNAJB6-related MFM was originally described as late-onset, slowly progressive limb-girdle weakness. However, several mutations can also cause an earlier, more severe, or distal-onset myopathy.53–56 In most cases, heterozygous mutations causing altered amino acids were determined to be localized on the G/F of the DNAJB6 protein. Variants in J domain causing distal myopathy with myofibrillar and rimmed-vacuolar pathology was also reported.57,58 P39 with the missense mutation c.206G > A on J domain manifested with late-onset and slowly progressive distal weakness similar as previous reports, and MFM-like pathology including obvious oxidative enzyme abnormalities on NADH-TR staining reflecting disorganized myofibrillar network, few amorphous materials and RVs. Although classified as unknown significance (VUS) according to the ACMG guidelines, it was predicted pathogenic in silico studies.
Pathological and clinical features in different genotype depended on the locations of mutations. For desminopathy, patients carrying mutations in the 1B and tail domains appear to develop early-onset and more severe cardiac disease compared to patients with mutations in the 2B segment. 51 Peripheral neuropathy appeared in patients harbored mutations in DES-tail domain. For filaminopathy, mutations in Ig-like rod domain are related to pathologically myofibrillar myopathy. These correlations are likely because each domain has its own function in stabilizing myofibrillar structures. 59 Giant axon nerve fibers, combined with peripheral neuropathy and spine rigidity could suggest the diagnosis of BAG3-related MFM.43,44 Limited by the small samples, the correlation between the genetic spectrum and clinical features of MFM patients was controversial. The genetic background, modifying genes, and other interacting proteins of MFM causative genes might contribute to the complexity of the phenotype-genotype relationship.
DES is the most common MFM causative gene in the present cohort of Chinese patients, followed by FLNC, BAG3 and TTN. The combination of NGS and pathological examination could greatly increase the diagnostic yield in MFM patients. While the novel variants we found were based on the precondition of MFM-like pathological features. The further functional study and detailed clinical data were needed to support the pathogenicity of novel variants.
Supplemental Material
sj-xlsx-1-jnd-10.1177_22143602241289220 - Supplemental material for Mutational and clinical spectrum of myofibrillar myopathy in one center from China
Supplemental material, sj-xlsx-1-jnd-10.1177_22143602241289220 for Mutational and clinical spectrum of myofibrillar myopathy in one center from China by Qi Wang, Peng Sun, Meng Yu, Zhiying Xie, Jiaxi Yu, Xiujuan Liu, Daojun Hong, He Lv, Jianwen Deng, Yun Yuan, Zhaoxia Wang and Wei Zhang in Journal of Neuromuscular Diseases
Footnotes
Acknowledgments
We would like to thank the patients and their family members for their participation in this study. This study was approved by the Local Ethics Committee of Peking University First Hospital.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National High Level Hospital Clinical Research Funding (High Quality Clinical Research Project of Peking University First Hospital) 2023HQ03.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are openly available.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.