
Abstract
Background:
The accurate diagnosis of titin-related myopathies (TTN-RM) is challenging due to the “gigantism” of the coding gene TTN with an incompletely understood landscape of normal genetic variation, an increasing number of pathogenic variants, and wide phenotypic variability of both cardiac and muscle involvement. Particularly in situations of potentially incomplete genotypes, clinicians need more phenotyping tools to help confidently determine the pathogenicity of variants in TTN and accurately diagnose titinopathies.
Objective:
To illustrate the pattern of muscle involvement found by muscle imaging in patients with TTN-RM.
Methods:
We reviewed the clinical and imaging data of patients with TTN-RM. Cross secitonal MR images of the lower extremity muscles were scored for degree of abnormality using the Mercuri scoring system and patterns were identified with comparison across muscle groups. Ultrasound images were also reviewed and described.
Results:
Eleven patients with TTN-RM had clinical and imaging data available for review. The relatively more severe involvement of the semitendinosus muscle in the hamstring group (“semitendinosus sign”) emerged as a consistent feature in patients with recessive TTN-RM despite clinical heterogeneity.
Conclusions:
Here we find that despite considerable complexity, the pattern of muscle involvement on MRI and ultrasound may aid in the confirmation of TTN-RM by establishing compatibility with the diagnosis.
Introduction
The TTN gene (2q31) (OMIM #188840) consists of the most number of reported exons (363) among human genes, and encodes the largest known human protein called Titin. 1 Titin is one of the main sarcomeric proteins, spanning approximately half of each sarcomere, extending from the Z-disc to the M-band. 2 Titin is expressed primarily in cardiac and skeletal muscle and plays a crucial role in the assembly, architectural foundation, cells signaling, elasticity, and force transmission of striated muscle.3,4
Advances in, and increased access to, massively parallel sequencing (MPS) have greatly facilitated the ability to screen for variants in large genes such as TTN in patients affected with a heterogeneous spectrum of disease. Heterozygous dominantly-acting TTN pathogenic variants are a common cause of familial and sporadic isolated dilated cardiomyopathy (DCM).5–7 More recently, pathogenic variants in TTN have been reported to cause a wide range of neuromuscular phenotypes, collectively known as titinopathies or TTN-related myopathies (TTN-RMs). The first pathogenic variant reported to cause TTN-RM was a Finnish founder 11 base pair insertion/deletion, manifesting clinically as adult-onset tibial muscular dystrophy (OMIM #600334) and reported in 2002. 8 Patients who were homozygous for this variant had a more severe clinical phenotype of childhood-onset limb girdle muscular dystrophy (LGMD) (OMIM #608807).8,9 Since that time, TTN variants have been reported as causing a wide spectrum of muscle disease with variable age of onset, severity and cardiac involvement; including Emery-Dreifuss muscular dystrophy, 10 congenital myopathy,11–13 myopathy with early respiratory failure,12,14–18 and adult onset distal myopathy with rimmed vacuoles, 19 Sahil myopathy and an early arthrogryposis phenotype. 13 In addition to the known clinical heterogeneity associated with causative variants in the TTN gene, the TTN histopathological spectrum is also diverse, including reports of internalized nuclei,20,21 minicores, 12 cytoplasmic bodies, 14 rimmed vacuoles, and myofibrillar abnormalities. 16
A confirmatory genetic diagnosis has become the gold standard in the evaluation of patients with hereditary neuromuscular diseases, and is essential for appropriate management, defining prognosis, accurate genetic counseling and determining candidacy for potential clinical trials. Despite this, the interpretation of genetic testing, particularly determining the relevance of variants of unknown significant in TTN, remains a challenge due to its large size, the alternative splice isoforms and reference sequences used, and the high degree of normal variation in the coding sequence. Therefore, careful analysis of variants using prediction models, in silico analysis tools, allele frequency, and familial segregation in the setting of the patient's clinical phenotype and muscle histotype is required. 22 To further complicate the diagnostic picture, the described clinical and histological features of tininopathies overlap considerably with other known myopathies and muscular dystrophies.
Muscle magnetic resonance imaging (MRI) has been used as a non-invasive tool to characterize the severity and patterns of muscle involvement in specific genetic muscle diseases. The recognition of distinct patterns of muscle involvement and sparing seen in genetically-defined muscle disease subtypes makes muscle imaging a useful phenotyping tool to narrow differential diagnoses and for assisting in the interpretation of variants of uncertain significance generated through MPS.23,24 For example, muscle imaging has proven to be helpful in patients with suspected collagen 6-related dystrophy (COL6-RD), for whom imaging commonly reveals a “central cloud” pattern, reflecting abnormal signaling around the central fascia of the rectus femoris.25,26 Additionally, involvement of the vastus lateralis with notable sparing of the rectus femoris can be suggestive of RYR1-related myopathies in the compatible clinical setting. 27 Furthermore, SELENON-related myopathies (formerly SEPN1), also known as rigid spine muscular dystrophy and multiminicore myopathy, are known to have more selective involvement of the sartorius muscle compared to the gracilis muscle and of the semimembranosus muscle, a unique feature that helps in differentiating these diseases from diagnostic mimickers. 28
Muscle imaging data for titinopathies have been presented in case series as part of phenotypic descriptions, but have not been evaluated in a systematic way as a disease marker.29–32 Here, we report phenotype and genotype data on a heterogeneous cohort of eleven patients with autosomal recessive TTN-RM and identify compatible characteristic imaging patterns to aid in the diagnostic evaluation and genetic confirmation of patients with a suspected titinopathy.
Materials and methods
Patients
Patients 2–11 were evaluated under protocol 12-N-0095 approved by the National Institute of Neurological Disease and Stroke Institutional Review Board. Patients were clinically evaluated at the NIH clinical center. Written informed consent and age-appropriate assent was obtained by a qualified investigator. Medical history was obtained, clinical evaluations and imaging studies were performed as part of the standard neurologic evaluation. DNA was obtained based on standard procedures. Clinical information and imaging studies on Patient 1 (P1) was provided by the patient's local neurologist. Patients with confirmed or suspected TTN-myopathy who could not undergo muscle MRI imaging were excluded.
Exome sequencing
Exome sequencing (ES) was performed on patients 2–5 and 10–11 at the NIH Intramural Sequencing Center (NISC) using Illumina's TruSeq Exome Enrichment Kit and Illumina HiSeq 2000 sequencing instruments. Results were confirmed with Sanger sequencing on an ABI 3130 × 1 capillary sequencer, in forward and reverse direction. Parental segregation was performed in all patients if possible. Variants were analyzed using Varsifter and seqr, and searched for in dbSNP, NHLBI EVS, and Exome Aggregation Consortium (ExAC Browser). 33 Patients 6 and 7 have previously been reported as 1044–1 and 1093–1, respectively. 20 Patient 8 was diagnosed with a commercial cardiomyopathy gene panel.
Muscle imaging
Muscle MRI data was collected retrospectively for all patients. For patients 2–11, muscle MRI was performed using conventional T1 weighted spin echo on a 1.5-T Achieva Phillips system or a 3.0-T Verio Siemens system. Non-contrast images were obtained from pelvis, thighs and legs in the axial plane. Slices were 8 mm thick and the gaps between slices were 10 mm. MRI images of patient 1 were transferred from the patient's home institution with appropriate consent.
Muscle scoring
Signal intensity was scored using a 5-point scale that was previously reported by Mercuri et al.,24,27 with the following classification:
0=normal 1=mild with only traces of increased signal intensity 2=moderate with increased signal in less than 50% of affected muscle 3=severe with increased signal intensity in more than 50% of the affected muscle 4=entire muscle replaced by abnormal signal
Median scores for each muscle were calculated for comparison between muscle groups. Scores were also summed within each patient at the proximal thigh, distal thigh and lower leg for correlation with clinical symptoms. After agreeing on the boundaries of each muscle (segmentation), three study team members scored T1-weighted MRI images of the proximal thigh, distal thigh, and lower leg muscles in a blinded fashion. MRI images from patients with other neuromuscular disorders were mixed in with the TTN-related muscle disease patients. Scored muscles included: Proximal thigh: rectus femoris, vastus lateralis, vastus intermedius, vastus medialis, sartorius, gracilis, adductor longus, adductor magnus, semimembranosus, semitendinosus, biceps femoris. Distal thigh: rectus femoris, vastus lateralis, vastus intermedius, vastus medialis, sartorius, gracilis, semimembranosus, semitendinosus, biceps femoris. Lower leg: medial and lateral gastrocnemius, soleus, tibialis anterior, tibialis posterior, extensor digitorum longus, peroneal muscle group. A final score was allocated based on two out of three scores being in agreement.
Muscle ultrasound
Ultrasound of skeletal muscles was performed on patients 2, 3, 7, 9, and 11 using a Siemens Acuson S2000 machine.
Results
Patients
A total of 11 patients with TTN-related muscle disease, 5 males and 6 females, ages 5–41 years old at the time of evaluation, had MRI imaging available for review. Clinical phenotype and genotype findings are summarized in Table 1. All patients had early onset muscle disease, nine presented with symptoms at birth or soon after, while for two patients childhood onset of symptoms was reported. Eight patients had delayed motor milestones. All patients experienced proximal muscle weakness, and 7 patients also had distal muscle involvement. Four patients had axial weakness with predominant neck flexion weakness; 7 patients had some degree of facial weakness as well. Seven patients were ambulatory, and 4 patients never achieved independent ambulation. Disease severity ranged from patients who had severe congenital hypotonia and never achieved the ability to walk (e.g., P4, P5, P7, P9) to those with relatively normal motor development and developing difficulty with stairs and getting up from the floor in adulthood (e.g., P10, P11).
Clinical characteristics and TTN mutations.
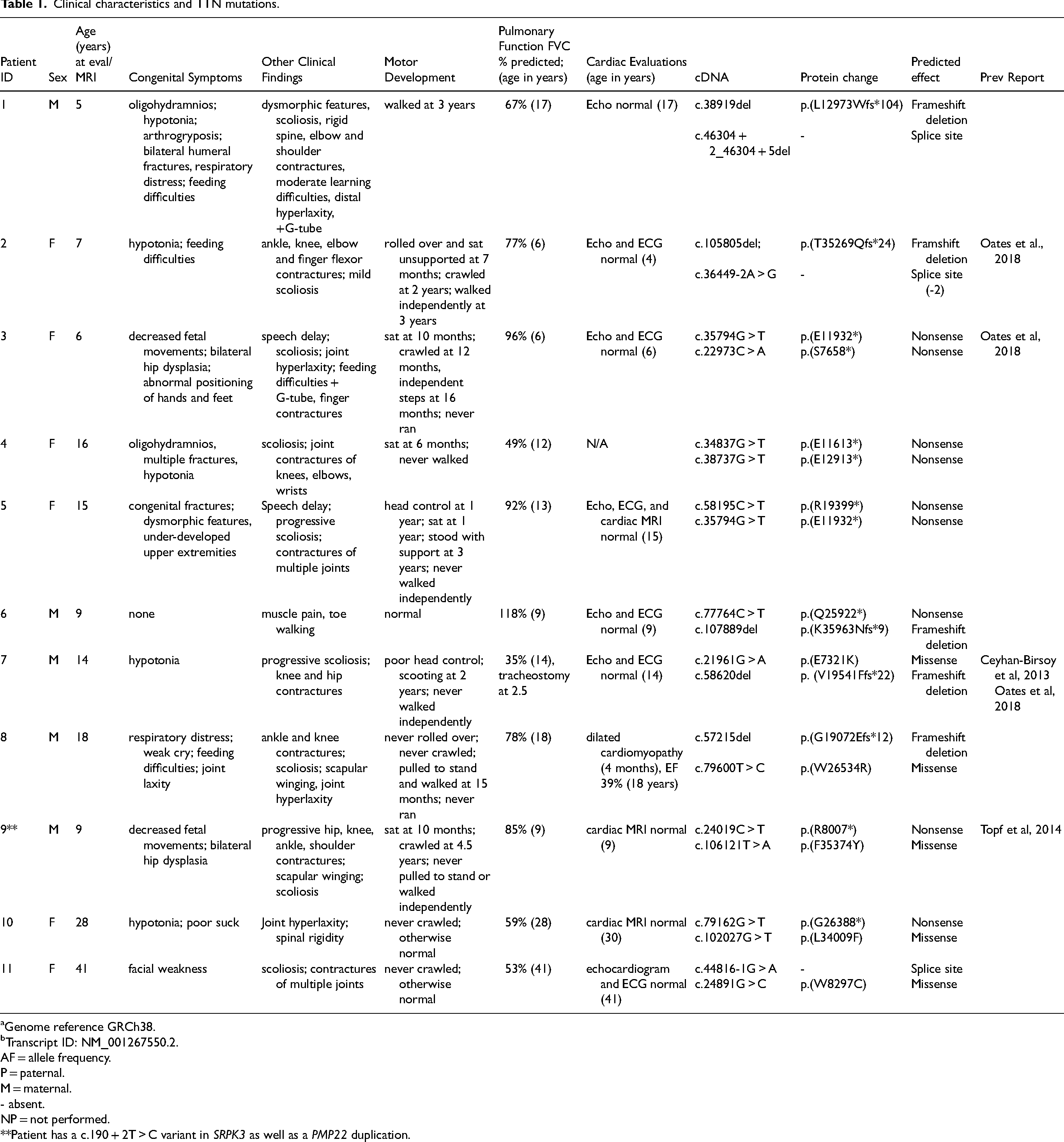
Genome reference GRCh38.
Transcript ID: NM_001267550.2.
AF = allele frequency.
P = paternal.
M = maternal.
- absent.
NP = not performed.
**Patient has a c.190 + 2T > C variant in SRPK3 as well as a PMP22 duplication.
One patient (Patient 7) had severe respiratory insufficiency (FVC 35%) and required ventilation via a tracheostomy, whereas the majority of patients had mild-to-moderate respiratory involvement (FVC < 60%). Cardiac evaluation was normal for all patients at the time of evaluation, except patient 8, who had dilated cardiomyopathy diagnosed at age 4 months. Creatine kinase (CK) values were largely within normal limits, ranging from 30 to 111 IU/L with the exception of Patient 6, who had CK elevation up to 2238 IU/L.
Variant analysis
All patients had recessive TTN-RM, with at least one truncating TTN pathogenic variant in biallelic heterozygosity with a second truncating variant (n = 4) or a rare missense variant predicted to be damaging (n = 7) (Table 1). Parental segregation testing was completed in all except for Patient 5 and 10 for whom paternal samples were not available. These variants were not reported in dbSNP, NHLBI EVS, and GnomAD databases. Patients 6 and 7 have been previously reported and evaluation of their variants included immunofluorescence analysis of muscle biopsies, splicing assays and gel electrophoresis of muscle proteins. 20
Muscle MRI pattern
The Mercuri scoring, as previously described, ranks muscle appearance qualitatively on a 5-point scale, based on the degree to which muscles are affected by degeneration and fatty-fibro replacement. 24 Following this methodology, higher scores in this scale reflect more advanced muscle degeneration.
The pattern of muscle involvement, with associated scoring, for each patient is illustrated in Figures 1 and 2. The hamstring group showed the highest score overall, indicating greater signal intensity, likely due to more extensive muscle degeneration and fibro-fatty change/replacement. The average scores of the posterior compartment of the thigh were 2.11 and 2.24, in the proximal and distal thigh respectively. In comparison, the average scores for the anterior thigh compartment were 1.38 proximally and 1.9 distally. The adductors were also relatively spared with an average score of 1.00 proximally. The median scores for each individual muscle are illustrated in Figure 2. Overall, our cohort demonstrates a notable selective or relatively more severe involvement of the semitendinosus which was present regardless of clinical severity or age at time of MRI. In the proximal thigh where this was most evident, the semitendinosus scored at least one point higher than its neighboring hamstring muscles in 9 out of the 11 patients.
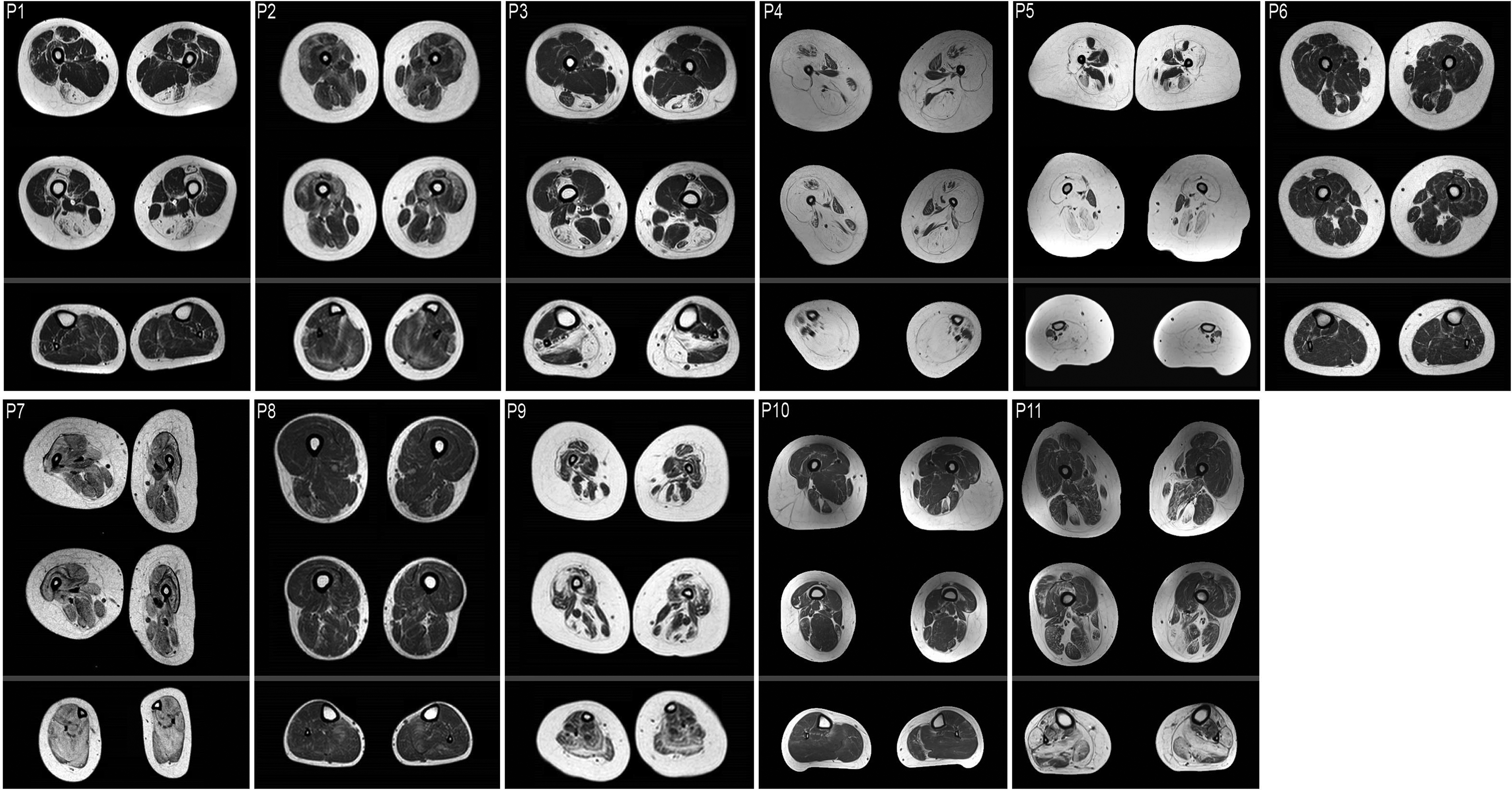
Selected axial T1-weighted MRI images of the lower extremities. These images were obtained for patients 1-11 at the following years-of-age, respectfully: 5, 7, 6, 16, 15, 9, 14, 18, 9, 28, 41.
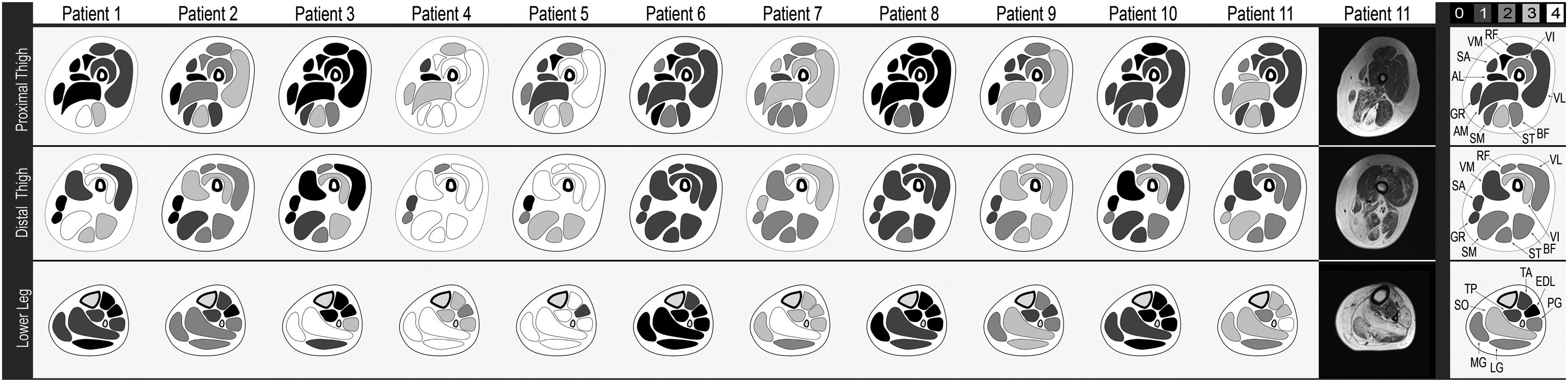
Muscle MRI pattern schematic. Schematic representations of axial T1-weighted muscle MRIs for Patients 1–11. Each schematic is shaded according to the score of each muscle, based on the key provided, at the proximal thigh, distal thigh, and lower leg. The far-right column illustrates the cumulative median scores for each muscle based on the eleven patients described. RF = rectus femoris, VL = vastus lateralis, VM = vastus medialis, VI = vastus intermedius, SA = Sartorius, GR = gracilis, AM = adductor magnus, AL = adductor longus, SM = semimembranosus, ST = semitendinosus, BF = biceps femoris, MG = medial gastrocnemius, LG = lateral gastrocnemius, SO = soleus, TA = tibialis anterior, TP = tibialis posterior, EDL = extensor digitorum longus, PG = peroneal group.
In the lower leg, the soleus had the greatest signal increase collectively. The peroneal group showed a greater signal increase as compared to the other anterior compartment muscles of the lower leg, particularly compared to the extensor digitorum longus, which is relatively spared.
Muscle ultrasound
The relative selective semitendinosus involvement that was observed on MRI imaging was also visualized by ultrasound as demonstrated in Figure 3. Similar to increased hyperintensity on MRI, increased echo intensity (whiteness) on ultrasound is reflective of increased fat and fibrous infiltration of the muscle. 23 Figure 3 demonstrates greater echo intensity of the semitendinosus as compared to neighboring semimembranosus and biceps femoris in patients 2, 3, 7, 9, and 11.
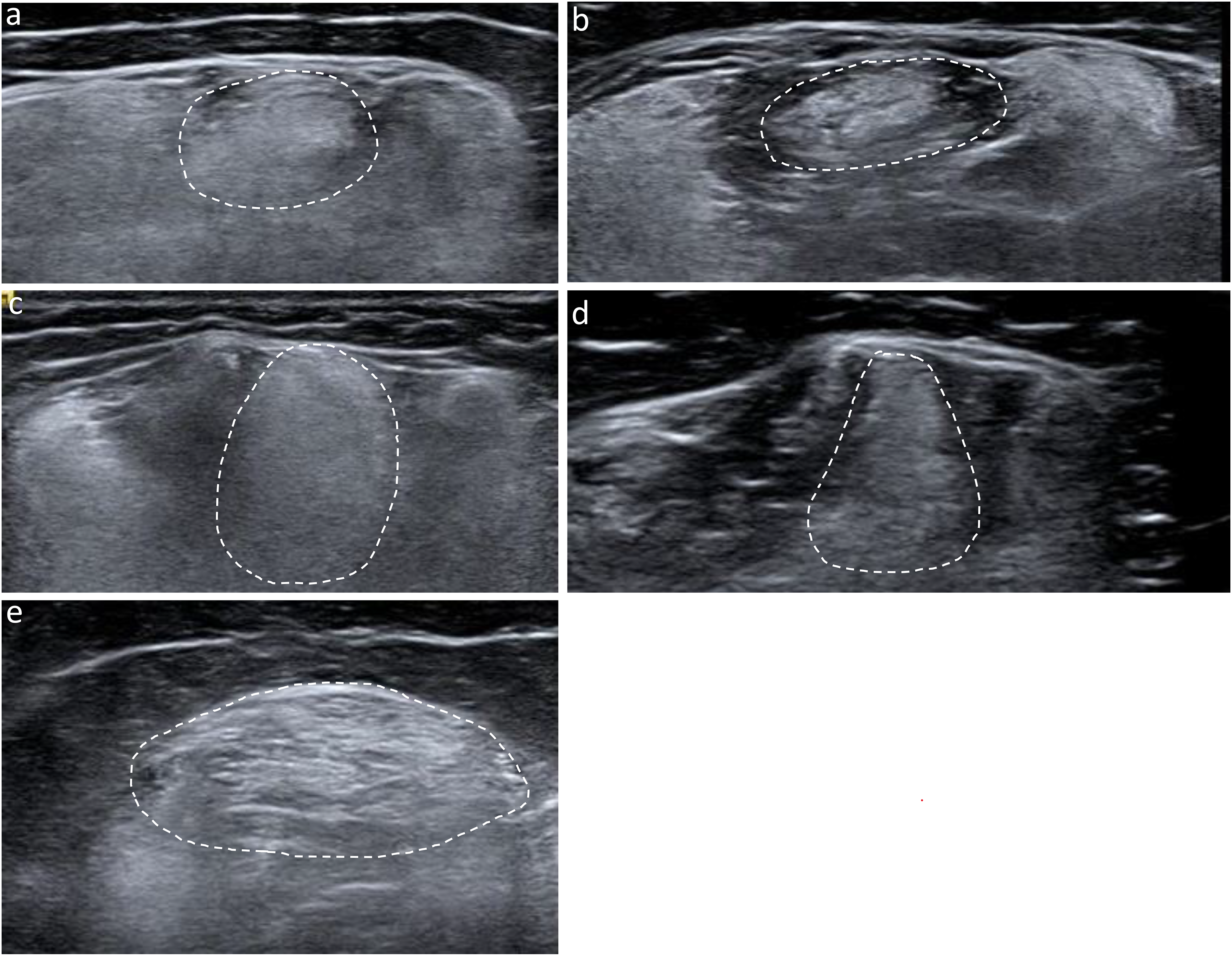
Ultrasound of the hamstring muscles. Normal muscles are typically dark whereas muscles with fibrosis and fatty infiltration have increased echogenicity (increased bright signal) as demonstrated in the semitendinosus muscles outlined by a dotted line. (a) Right hamstrings, patient 2 at 7 years old. (b) Left hamstrings, patient 3 at 6 years old, (c) Right hamstring, patient 7 at 13 years old. (d) Left hamstring, patient 9 at 5 years old. (e) Right hamstring, patient 11 at 41 years old.
Correlation between MRI and clinical findings
The patients who never achieved independent ambulation (patients 4, 5, 7, and 9) had the highest cumulative scores in muscles of the thigh and 4 of the 5 highest scores overall. Patient 11, the oldest patient in the cohort, had the 4th highest cumulative score. Of the patients who had normal motor development (6, 10, 11), patients 6 and 10 had the second and third lowest cumulative scores. The lowest cumulative score, indicating the most normal appearing muscle on MRI, was in patient 8, who had the most severe cardiac disease (Supplementary Table).
Discussion
TTN-RMs encompass a group of myopathies that are increasingly recognized as having significant clinical heterogeneity including congenital through adult-onset muscle diseases with both autosomal dominant and recessive modes of inheritance. The latter is the mode of inheritance to be expected in early onset TTN-RM and is considerably more common in the skeletal muscle phenotypes associated with TTN pathogenic variants. Recessive TTN-RM is associated with considerable phenotypic heterogeneity, as illustrated in our cohort. Achieving a complete genetic diagnosis can be challenging due to TTN's genetic “gigantism,” resulting in a large and increasing number of pathogenically unassigned variants. In such situations it is very helpful to create tools allowing for a more confident assignment of a given phenotype in a patient as TTN-compatible. Here we systematically analyze muscular imaging data to explore the use of muscle MRI as such a tool to aid clinicians in diagnosing TTN-RM with more confidence, particularly in the setting of inconclusive genotypes, such as missense variants of uncertain significance or a “missing second allele” scenario.
Patients in our cohort presented with congenital or early onset muscle disease; however, they experienced extensive variability in progression of muscle weakness, respiratory decline and cardiac involvement. The severity of weakness in our cohort ranged from mild motor disability to non-ambulation, including those who never achieved independent ambulation (Patients 4, 5, 7 and 9). Only one patient (patient 8) had cardiac involvement manifesting clinically as severe dilated cardiomyopathy at age 4 months. Interestingly, this patient had relatively mild skeletal muscle involvement on imaging, had only mildly delayed motor development, and was ambulatory. From a respiratory standpoint, again we saw a spectrum of involvement with one patient requiring tracheostomy in childhood (patient 7), while the majority of our patients were found to have mild-to-moderate respiratory insufficiency based on pulmonary function testing.
Despite the variability of clinical phenotype and severity of muscle involvement by imaging demonstrated in our patients, we identified a suggestive imaging finding shared among our patients: Patients with TTN-RM frequently have relatively more severe or selective involvement of the semitendinosus muscle, in comparison to surrounding muscles of the hamstring group, which was particularly evident in the proximal thigh. In our cohort of 11 patients, all but two showed this “semitendinosus sign”. Five patients had Mercuri scores that were 2 or more above the semimembranosus. Patient 4 had diffusely severe or “end stage” appearing muscle which prevented an accurate assessment of differential muscle involvement. The hamstring muscles in patient 7's MRI appeared equally affected. The semitendinosus sign was initially reported in patients with a dominantly inherited titinopathy with the HMERF phenotype.34,35 It has been further reported in small case reports of patients with recessive congenital titinopathies, but has not been highlighted as a characteristic feature.13,29,30,32 By utilizing a blinded scoring system, we were able to more objectively look for disproportionate semitendinosus involvement even when it may not be evident at first glance.
This hamstring pattern does appear to be present early on in TTN-RM and in cases with milder phenotypes, as demonstrated in our cohort. Muscle MRI performed in patients who are further along the course of their disease with evidence of severe muscle weakness may no longer reveal the patterns of muscle involvement that can be seen earlier in the disease. Muscle ultrasound may also be more sensitive to identifying mild and early symptoms and is more amenable for use in young children, while ultrasound sensitivity may decrease in patients at later stages of disease. Our study demonstrated that the pattern of involvement of the semitendinosus with relative sparing of the semimembranosus and biceps femoris muscles was also detectable on muscle ultrasound. Muscle ultrasound can also serve as an alternative imaging modality for those unable to safely obtain or tolerate an MRI.
The application of MRI excludes patients who have MRI incompatible cardiac and surgical devices and very young infants or children who may be unable to tolerate the procedure. This may have led to selection bias in our cohort and thus skewing our population towards older patients and those without cardiac involvement. It is also important to note that while selective involvement of the semitendinosus may have an association with TTN-RM based on this report and others, it is not exclusively found in TTN-RM. For example, it has been previously reported in myofibrillar myopathies, particularly due to DES variants.36–38 The clinical phenotype and histotype of patients with myofibrillar myopathy, who generally have a later onset myopathy, would be distinctive from that of a recessive TTN-RM, which typically presents in infancy with hypotonia, weakness and early contractures. Thus, like all other ancillary tests, muscle MRI findings need to be interpreted with close consideration of the patient's clinical phenotype.
Our data suggests that the semitendinosus sign can be observed both on muscle MRI and muscle ultrasound in TTN-RM and demonstrates that this imaging sign is fairly independent of the patient's clinical phenotypic severity or underlying recessive genotype. Within a compatible clinical and genetic context, the selective involvement of the semitendinosus offers additional supportive evidence of a TTN-RM, adding to a “compatibility matrix” of genotype, histotype, and extended phenotype including imaging. The pattern can thus aid in TTN variant interpretation and facilitate diagnosing a likely TTN-RM even if the genotype is still tentative. It is also helpful in distinguishing from diagnoses with clinical presentations with overlapping features and frequent variants of uncertain significance found on MPS, such as collagen-6 related muscular dystrophy. Limitations of this study include the small sample size and selection bias as all patients were part of a selective myopathy research study. Additionally, while all patients are felt to have clinical, histological and/or genetic evidence supporting the diagnosis of TTN-RM, some patients do have missense variants which must be interpreted with caution. A large prospective and longitudinal study would help to further establish the sensitivity and specificity of this “semitendinosus sign” in inherited neuromuscular disease, and specifically in TTN-RM.
Conclusions
We report on a cohort of 11 patients with recessive TTN-RM. Even though our patients had striking clinical heterogeneity, a common characteristic imaging finding of relatively more severe or selective involvement of the semitendinosus, was identified on muscle MRI and muscle ultrasound. This semitendinosus can therefore be used as an adjunctive tool to help with TTN variant interpretation and diagnosis in patients with clinical and histological features suggestive of TTN-RM.
Footnotes
Acknowledgements
The authors thank the patients and their families for participating, and Gilberto (Mike) Averion and, Christopher Mendoza for their help. We also thank the NIH Intramural Sequencing Center, Daniel MacArthur and Fengmei Zhao (Analytic and Translational Genetics Unit at Massachusetts General Hospital in collaboration with the Broad Institute of Harvard and Massachusetts Institute of Technology), the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at ![]() .
.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Bonnemann is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review. No other conflicts to declare.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by National Institutes of Health Intramural Research Program funding from the National Institute of Neurological Disorders and Stroke.
Data availability
Supplemental material
Supplemental material for this article is available online.