
Abstract
In cell biology research and pharmaceutical discovery, physiological responses of mammalian cells are commonly screened using transcriptional assays. Although firefly luciferase is widely used because of its rapid and simple assay, greater precision can be achieved using a second reporter as an internal control. Renilla luciferase serves as an efficient internal control because it can be measured as easily and rapidly using the same instrument. The Dual-Luciferase™ Reporter (DLR™) Assay developed at Promega measures both reporters sequentially within each sample, which eliminates the need to separate the test sample into aliquots for each assay. The expression of each reporter is independently quantitated using selective assay conditions based on their distinctive chemical characteristics. The firefly luciferase is initiated first by the addition of LAR II to the sample or cell lysate. Following measurement of the luminescent output, the firefly reaction is rapidly quenched and the Renilla reaction is simultaneously activated by addition of Stop & Glo™ Reagent, and the luminescence is measured a second time. The DLR™ Assay allows quantitation of both reporters within 4 seconds per well using a 96-well luminometer equipped with two reagent injectors. Multi-well plates may be processed even more rapidly using a CCD-based imaging system.
INTRODUCTION
Transcriptional assays may be used to study a broad range of cellular activities, such as receptor function, signal transduction, protein-protein interaction etc. (1). By coupling transcription to luciferase expression, these assays can be easily automated making them suitable for high throughput screening. The Dual-LuciferaseTM Reporter Assay enhances this capability further by also enabling assay of an internal control reporter in each experimental sample. In the DLR™ system, the firefly luciferase serves as the primary experimental reporter, while the Renilla luciferase serves as the control reporter. Implementation of an internal control for normalization of experimental results addresses problems that might arise from non-specific effects on gene expression, and therefore provides the means for more reliable data. This increased reliability is particularly valuable in screening applications, where the quantitative results from thousands of different compounds are evaluated. As an internal control, Renilla luciferase has similar assay characteristics to firefly luciferase, and it can compensate for physical variables (e.g., “edge effects” in multi-well plates) and physiological variables (e.g., cell viability and growth). Thus, by decreasing experimental error, the DLR™ Assay system can enable more accurate and dependable results with increased productivity.
METHOD
Cells:
Stable CHO cells lines were generated from CHO cells co-transfected with pCIneo vectors containing the firefly and Renilla gene separately. Hygromycin B was added to the medium for selection 2 days after transaction. The resistant cells were cloned independently and checked for both enzyme activities. Clones that had the highest levels of activity for both enzymes were selected, and then maintained in medium containing Hygromycin B.
In addition CHO cells were transiently transfected with both pGL3-Control and pRL-SV40 for preparation of lysate used in Figure 1. In general cells were plated at 3×104 cells per well in a 96-well plate.
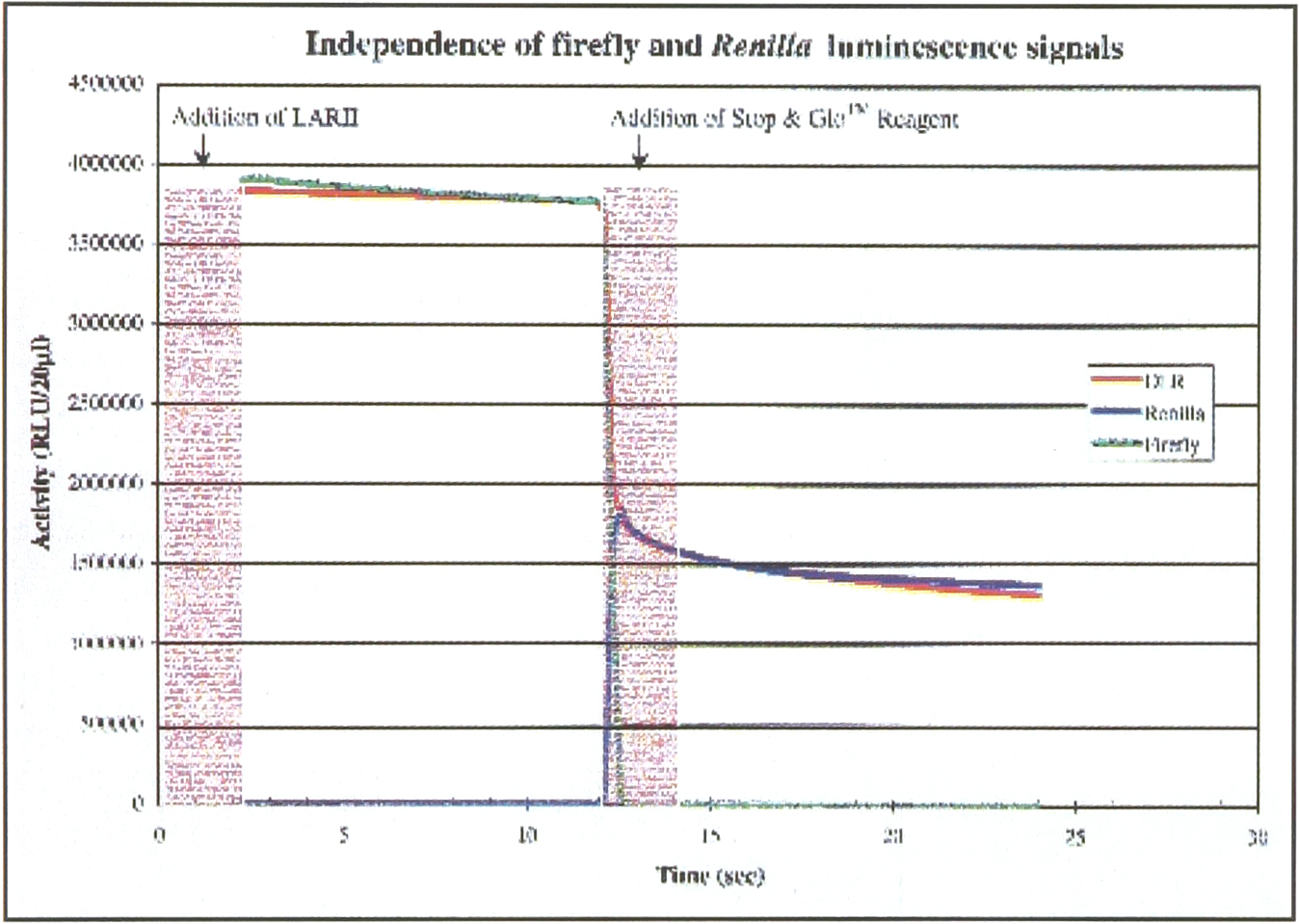
Cell lysate was prepared in PLB from CHO cells transiently co-transfected with both firefly and Renilla luciferase DNA. The activities for both luciferases were measured using the standard DLRTM Assay (red), DLRTM Assay without luciferin (blue) and DLRTM Assay without coelentrazine (green). Luciferase Assay Reagent II (with/without luciferin) was added at 0 time, and Stp & GloTM Reagent (with/without coelentrazine) was added at 12 second. Each part of the assay was measured with a 2 second pre-read delay (shaded area) followed by a 10 second read time. The assay was performed in a 96-well plate.
Cell lysate:
Cell lysates were prepared either directly in multi-well plates or transferred into the wells from another source. In either case Passive Lysis Buffer (PLB) was used, which as the name suggests will lyse cells without the requirement for scraping and/or a freeze/thaw step.
ASSAY FOR BOTH REPORTERS:
To 20ml cell lysate, is added a 100ml of Luciferase Assay Reagent II. After measuring the luminescence generated, 100ml of Stop & Glo™ Reagent is added to the same well and another measurement is taken. A 96-well plate reading luminometer equipped with two injectors was utilized. Except where indicated, the settings incorporated a 2 second pre-read delay followed by a 10 second read time after the addition of each reagent.
RESULTS & DISCUSSION
Reaction kinetics for both luciferases in the DLR™ assay can be seen in Figure 1. The luminescence from firefly luciferase is activated by adding the first assay reagent (Luciferase Assay Reagent II). After measuring luminescence, the first reaction is quenched and Renilla luciferase activity is simultaneously activated by addition of the second reagent (Stop & Glo™). Luminescence from Renilla luciferase is then measured without interference from firefly luciferase activity.
To demonstrate the independence of firefly and Renilla luminescence signals, the following two changes to the DLR™ method were made (Figure 1). First, using the same enzyme preparation and reagents, the DLR™ assay was performed without firefly luciferin in Luciferase Assay Reagent II. The figure demonstrates that, no activity from firefly luciferase was detected in the absence of luciferin, the luminescence from Renilla luciferase was unchanged. Second, the DLR™ assay was done without coelenterazine in the Stop & Glo™ reagent (the substrate required for Renilla luciferase). In this case, no Renilla luciferase activity was detected, while firefly luciferase activity remained unchanged. It is clear from these signal traces that the complete DLR™ assay constitutes sequential, independent assays of the firefly and Renilla luciferases.
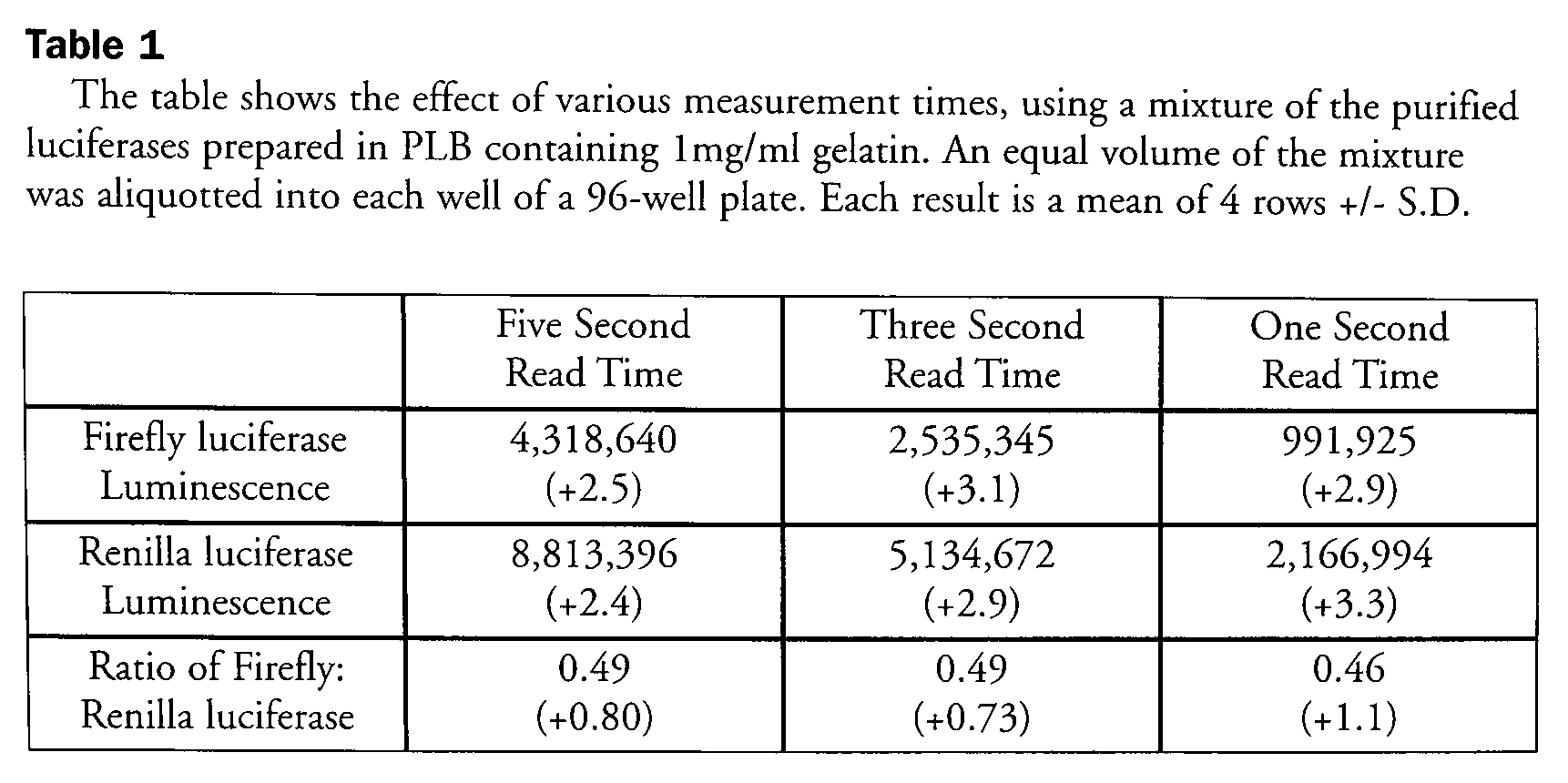
This reaction also illustrates that quenching of the firefly enzyme reaction and activation of the Renilla luciferase reaction occurs within less than 0.5 seconds. This quick interchange between the two different luminescence signals is an opportunity for reducing the delay time required between the assays to less than 1 second. In a similar fashion, the read time for each luciferase can also be reduced to 1 second, thus increasing overall assay throughput. This flexibility in the assay time is possible since the signals are relatively steady for several seconds. Table 1 shows the effect of read times on the DLR™ assay. Even though the luminescence values of each luciferase change with the different read times, the normalized ratio of the luminescence is independent of the read times. Note also that even when using purified enzymes, the relative errors decrease about 3-fold after normalization.
The table shows the effect of various measurement times, using a mixture of the purified luciferases prepared in PLB containing 1 mg/ml gelatin. An equal volume of the mixture was aliquotted into each well of a 96-well plate. Each result is a mean of 4 rows +/- S.D.
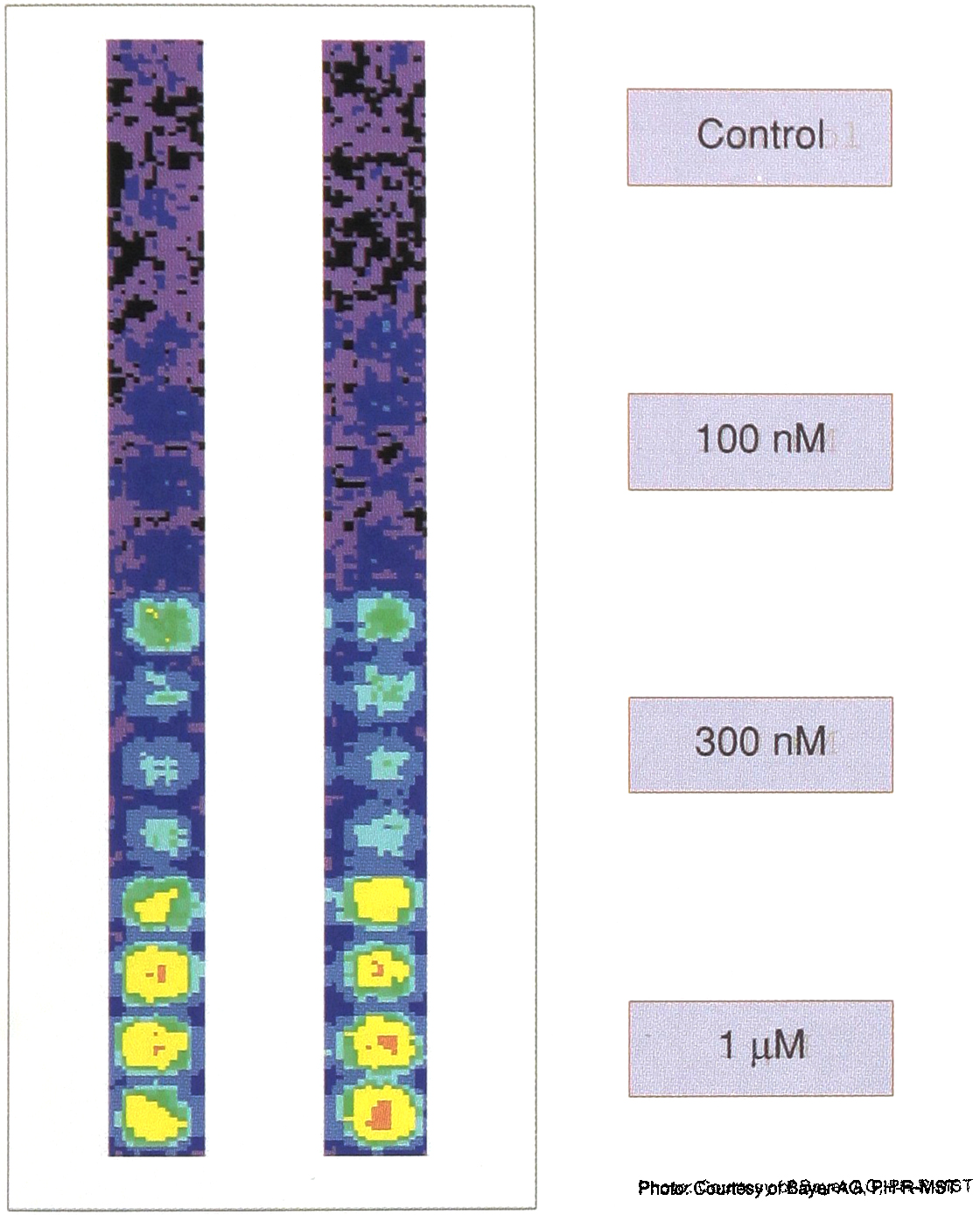
Titration of the firefly and Renilla luciferases shows that the linear range extends over seven orders of magnitude (4), and the sensitivity is at least 10–20 moles for firefly luciferase and 10–18 moles for Renilla luciferase (Figure 2). In the latter case, the departure from linearity below 10–18 moles is due to autoluminescence by coelentrazine. The Dual-Luciferase™ Reporter Assay therefore provides a very wide working range even at low attomole levels of both reporters.
The assay was performed with a mixture of firefly and Renilla luciferases prepared in PLB containing 1mg/ml gelatin. A titration was performed by diluting the stock mixture of both enzymes, and measuring activities in a 96-well plate.
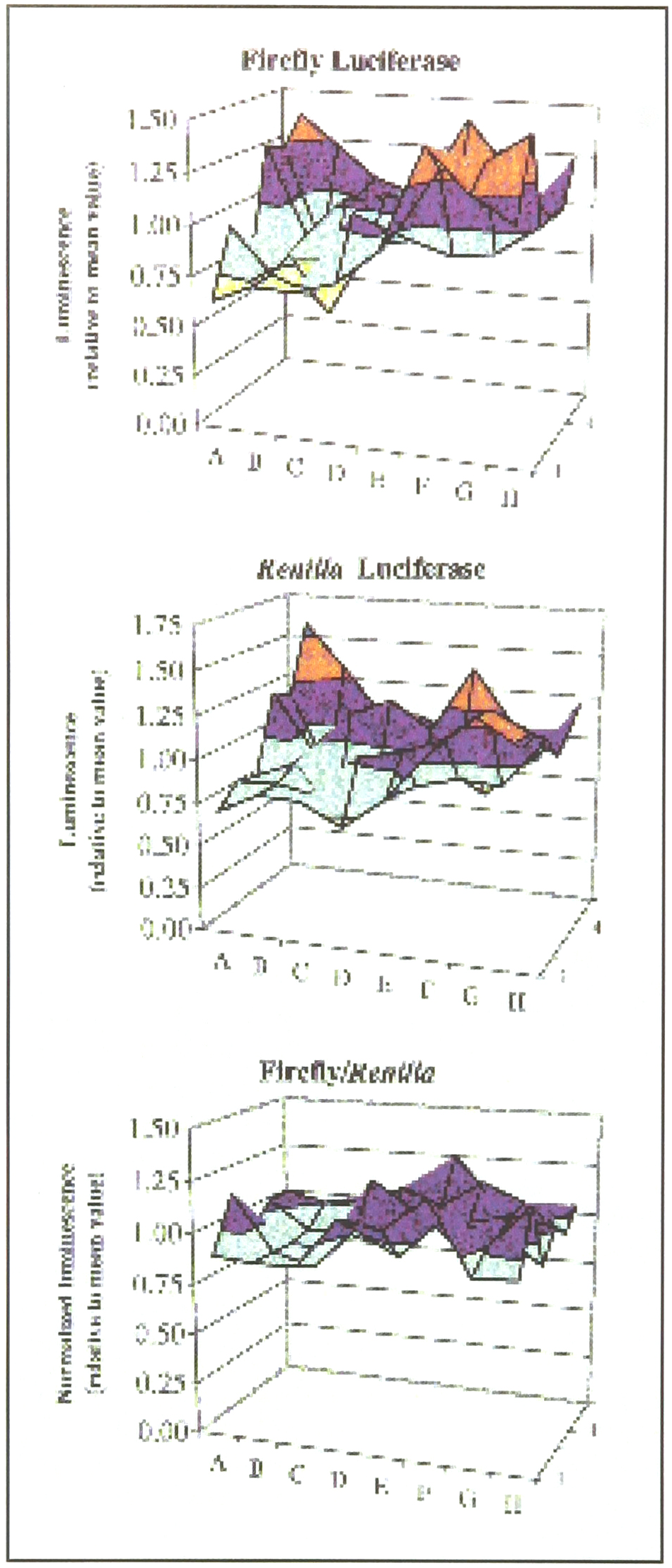
The ability of the DLR™ assay to correct for edge effects is demonstrated in Figure 3. In spite of using stably co-transfected CHO cells, there was considerable variation across the 96-well plate when only firefly luciferase or Renilla luciferase activity was measured. However, when firefly luciferase activity was normalized with Renilla luciferase activity then a more uniform pattern of expression was seen across the plate. In this case, edge effects, were seen despite using uniform plating conditions and a single pool of cells. Thus again, even under nearly ideal conditions an internal control is shown to decrease the relative error between samples.
The DLRTM Assay was performed in a 96-well plate on CHO cells stably transfected with both firefly and Renilla luciferase genes. The cells were washed with PBS and then lysed in situ with 20ml of Passive Lysis Buffer. Following a 10 minute wait period for completion of cell lysis, each well was assayed for both firefly and Renilla luciferase activity. Panels A and B indicate individual luminescence of firefly and Renilla luciferases respectively per well. The values shown are relative to the mean value, which is set at 1.0. Panel C represents the ratio of firefly luciferase activity normalized to Renilla luciferase within each well.
CONCLUSION
The DLR™ assay enables reporter measurements with an internal control that are simple, fast and reliable. This assay is suited for screening applications where large amounts of reliable quantitative data are required. The total assay time can be reduced considerably by shortening the read time without compromising results. In addition these times can be further reduced with the use of a cooled CCD camera instead of a luminometer (5). The assay, because of its characteristics and presence of an internal control, can reduce relative experimental errors and compensate for variable experimental conditions. This could reduce the number of false results and the number of repetitions required for an experimental trial.