
Abstract
The hepatitis C virus (HCV) infects more than 200 million people globally, with increasing incidence, especially in developing countries. HCV infection frequently progresses to chronic liver disease, creating a heavy economic burden on resource-poor countries and lowering patient quality of life. Effective HCV diagnosis, treatment selection, and treatment monitoring are important in stopping disease progression. Serological assays, which detect anti-HCV antibodies in the patient after seroconversion, are used for initial HCV diagnosis. Qualitative and quantitative molecular assays are used to confirm initial diagnosis, determine viral load, and genotype the dominant strain. Viral load and genotype information are used to guide appropriate treatment. Various other biomarker assays are performed to assess liver function and enable disease staging. Most of these diagnostic methods are mature and routinely used in high-resource countries with well-developed laboratory infrastructure. Few technologies, however, are available that address the needs of low-resource areas with high HCV prevalence, such as Africa and Southeast Asia.
Introduction
The hepatitis C virus (HCV) is the most common blood-borne pathogen. Globally, more than 200 million people are infected with HCV, predominately in Asia and Northern Africa ( Figure 1 ). 1 Approximately 80% of infected individuals develop chronic HCV infection, 2 and of this nearly 20% develop liver complications such as fibrosis, hepatocellular carcinoma, cirrhosis, and end-stage liver disease. 3 More than 50% of HCV cases remain asymptomatic in the initial phase, and are not diagnosed prior to onset of severe hepatic fibrosis. 4 HCV is therefore the leading cause of liver transplants in Europe and America.3,5 Early detection of HCV and appropriate therapeutic management in chronic patients can prevent these complications and therefore improve patient health and reduce patient morbidity and mortality.
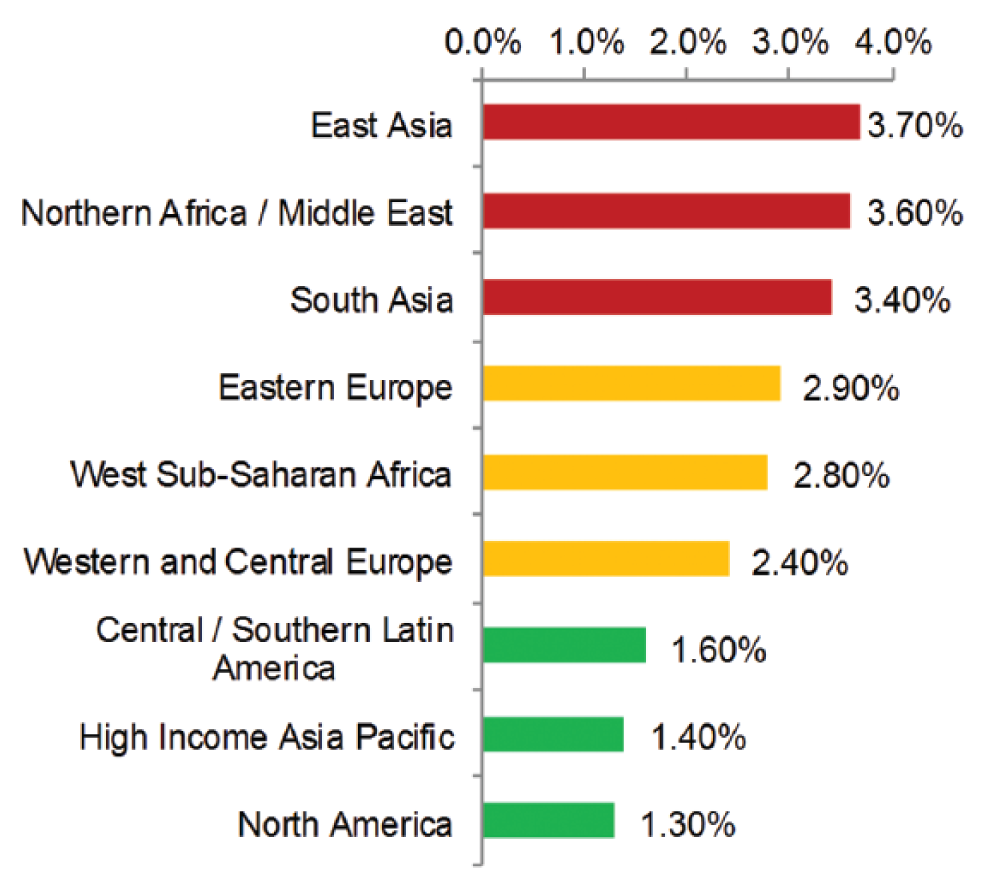
Hepatitis C virus (HCV) prevalence in selected regions. High, medium, and low HCV burden regions are depicted in red, orange, and green, respectively. Data from Mohd Hanafiah et al. 6
HCV is transmitted through percutaneous exposure 7 (e.g., due to sharing needles during recreational drug use and inadequate infection control in health care units). Population groups at risk for HCV infection include intravenous drug users, human immunodeficiency virus/acquired immune deficiency syndrome (HIV/AIDS) patients, children born by HCV-infected mothers, and individuals who received blood transfusions or organ transplants before rigorous screening protocols were implemented. Furthermore, as a result of disproportionately high HCV prevalence in persons born between the years 1945 and 1965 (comprising approximately 75% of all HCV infections), the Centers for Disease Control and Prevention (CDC) has implemented a new guidance in 2012 recommending mandatory one-time screenings for all persons born within the target years. 8
HCV is an enveloped ribonucleic acid (RNA) virus with 10 protein coding genes, which targets liver hepatocytes. The viral proteins elicit an immune response, which leads to the activation of cytotoxic CD4 T lymphocytes. In an attempt to stop further infection, these cytotoxic T cells seek out and lyse infected hepatocytes that display viral proteins on their surface. This host immune response is thought to lead to cirrhosis of the liver and eventually to hepatocellular carcinoma. 9
Diagnosis and Treatment of HCV
HCV diagnosis, treatment selection, and treatment monitoring involve a variety of biomarkers and diagnostic testing methods ( Fig. 2 ).
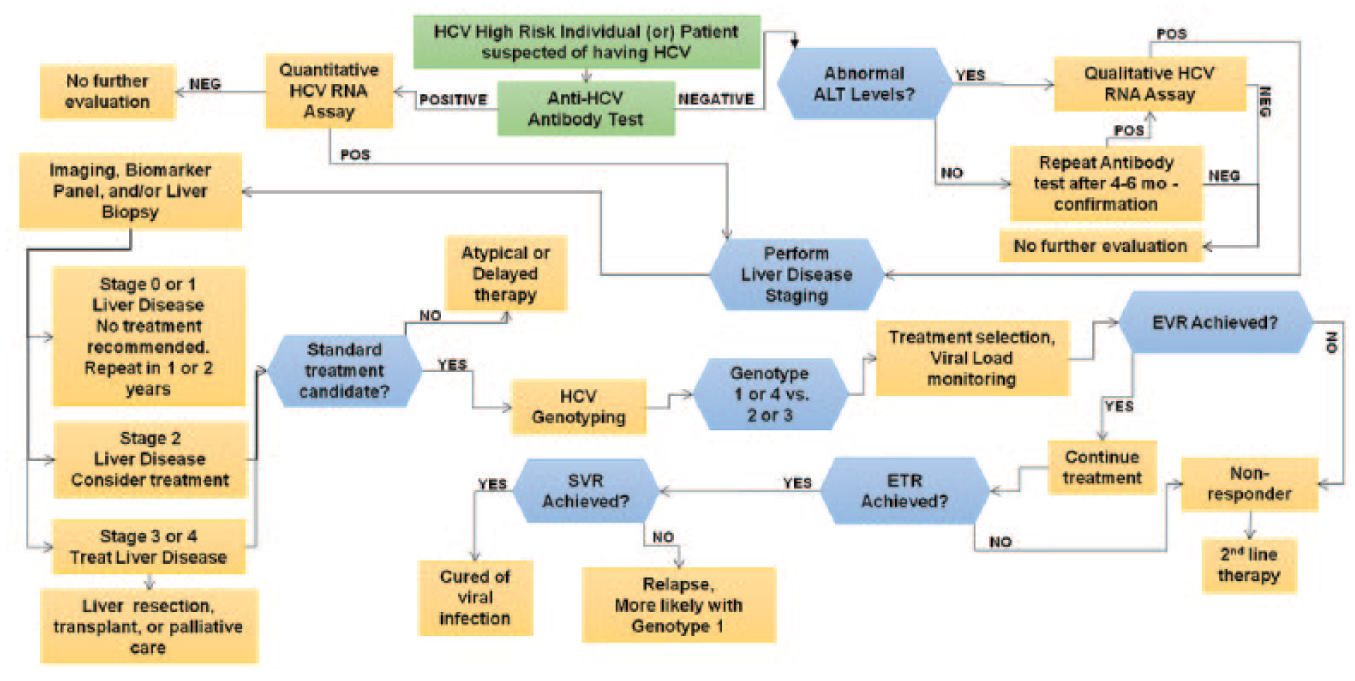
The CDC recommends periodic HCV screening for all individuals in high-risk groups. In addition, since 2012, one-time screening is recommended for all persons born between 1945 and 1965.8,13 The initial test for screening and diagnosis of HCV recommended worldwide is the serological enzyme immunoassay (EIA), which detects anti-HCV antibodies in the individual. In addition, the serum level of alanine transaminase (ALT), a liver enzyme, is a common biomarker used for initial assessment of liver damage ( Table 1 ). 14 Initial diagnosis based on serology or elevated ALT levels is usually confirmed via highly sensitive molecular diagnostic assays that detect the genomic RNA of the virus from the serum or plasma of the patient. Patients with confirmed HCV infection undergo disease staging to assess the extent of liver damage and to decide if the patient is a candidate for treatment.10–12 Histopathological examination of a liver biopsy sample, with standardized scoring via the METAVIR or the Knodell scoring system, 15 is commonly used to determine the disease stage. These methods provide information about liver inflammation and scarring, which are hallmarks of disease progression, enabling physicians to gauge the aggressiveness of the HCV infection. 16 Liver biopsy is expensive and highly invasive, however, and it affects patients mentally and physically. 3 Ultrasound imaging–based transient elastography can be used to assess the liver stiffness. 17 This method can diagnose Stage 4 liver cirrhosis with good accuracy, but is moderately accurate in detecting and differentiating earlier stages of liver fibrosis (Stages 1–3). 18 Furthermore, the results can be confounded in obese patients. As an alternative, minimally invasive biomarker tests are increasingly used to stage the disease ( Table 1 ). HCV patients with advanced liver cirrhosis or liver cancer may require liver resection or transplantation, which is highly invasive and has a low overall success rate.19,20 Therefore, the goal is to initiate treatment early on during disease progression (Stage 2) to halt viral replication and prevent liver damage.
Biomarkers for Liver Disease Diagnosis and Staging.
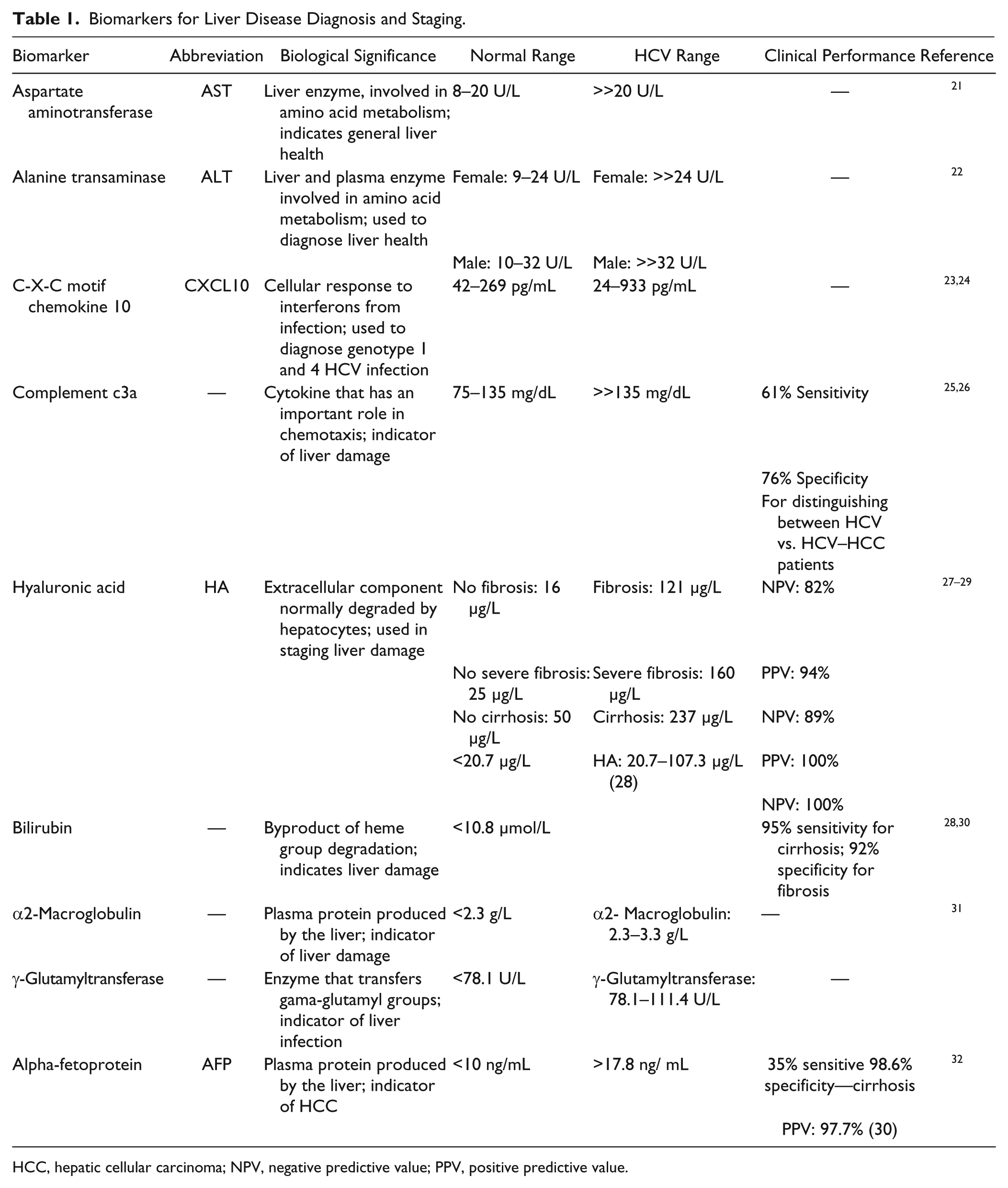
HCC, hepatic cellular carcinoma; NPV, negative predictive value; PPV, positive predictive value.
Once a patient has been identified as a candidate for treatment, the physician must select the appropriate therapy. 33 Treatment options to control and suppress viral replication include stimulating the host’s innate immune response with pegylated interferon (peginterferon) alpha to slow viral spread, 7 or HCV-directed therapeutics (e.g., the nucleoside analog ribavirin) that interfere with viral replication or maturation. 34 The most effective HCV treatment regimen depends on the genotype of the predominant viral strain infecting the patient. Therefore, HCV genotyping is commonly performed prior to treatment initiation. Overall, there are 11 HCV genotypes, further divided into subtypes and strains, with genotypes 1–6 being the most common. 35 Genotype distribution varies significantly by country and region ( Table 2 ). For example, HCV genotype 1 is responsible for about 60% of HCV infections in the United States, 36 genotype 2 accounts for >50% of infections in China, genotype 3 causes more than 76% of HCV infections in India, 37 and genotype 5 is highly prevalent in South Africa. 38
HCV Genotype Prevalence in Selected Countries (%).
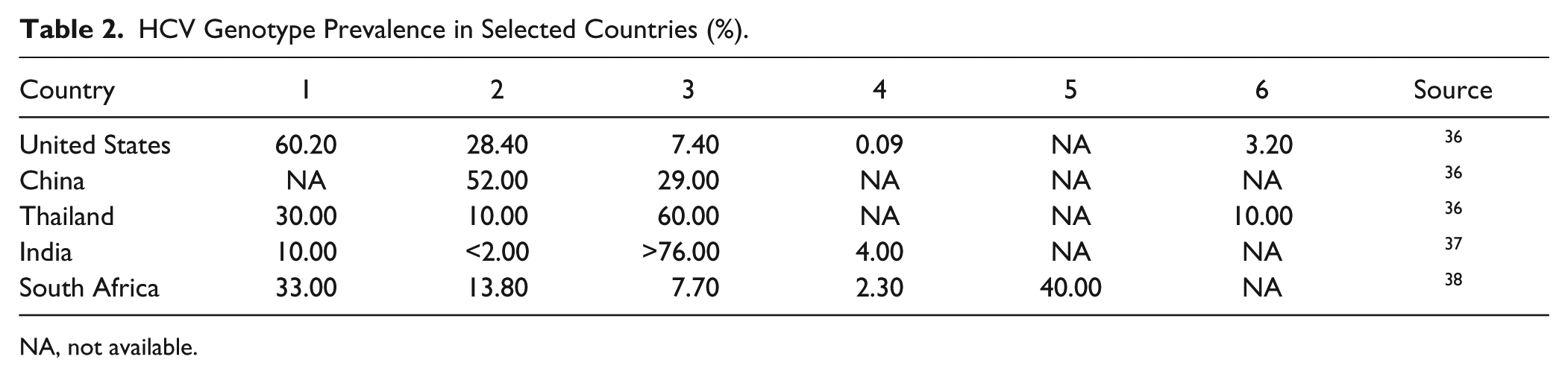
NA, not available.
The HCV genotype allows for proper drug selection, dosage, and overall treatment duration ( Table 3 ). In general, patients infected with HCV genotypes 1, 4, and 6 require more aggressive treatment for a longer duration, whereas HCV genotypes 2 and 3 respond more favorably to therapy. Traditionally, patients have been treated via a combination therapy of peginterferon alpha and ribavirin. Peginterferon must be injected, can cause significant side effects, and has low efficacy as monotherapy, especially for HCV genotype 1. 39 The efficacy of ribavirin has been undermined by increasing viral resistance. 40 Therefore, treatment may be augmented with second-line drugs such as telaprevir and boceprevir, which target the viral nonstructural proteins 3 and 4 (NS3 and NS4). 41 Miravirsen, a micro-RNA that blocks viral replication, is in clinical trials as novel second-line drugs to manage HCV infection. 42 Cost-effectiveness studies in low-income countries conducted specifically in both Egypt and Vietnam have shown that higher treatment rates have a greater population impact; therefore, treatment of later-staged patients is highly beneficial.43,44
Different HCV Genotypes and Their Most Effective Treatment Regimens. 11
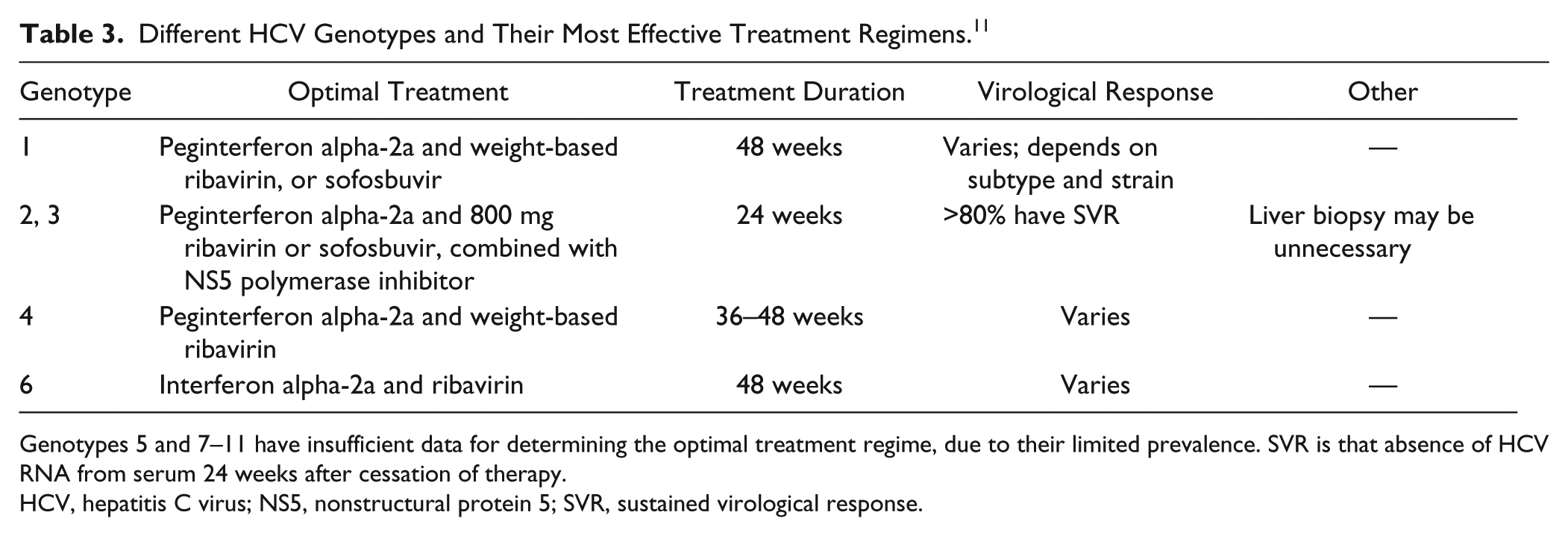
Genotypes 5 and 7–11 have insufficient data for determining the optimal treatment regime, due to their limited prevalence. SVR is that absence of HCV RNA from serum 24 weeks after cessation of therapy.
HCV, hepatitis C virus; NS5, nonstructural protein 5; SVR, sustained virological response.
The RNA polymerase inhibitor sofosbuvir (Sovaldi), a nucleoside analog developed by Gilead and approved by the US Food and Drug Administration (FDA) in 2013, has the potential to significantly change HCV treatment. Sofosbuvir can be administered orally and has demonstrated high efficacy. During clinical trials, sustained virological response (SVR), a surrogate endpoint for HCV cure, was observed in >90% of HCV genotype 2 treatment-naïve patients, and in ~71% of patients who previously failed to achieve an SVR using interferon-based therapies. 45 Sofosbuvir is also the first drug shown to be effective without interferon gamma co-administration. At the time this article was written, sofosbuvir has been approved for treatment of HCV genotypes 2 and 3, with clinical trials for other genotypes ongoing. The recommended 12-week treatment regimen can, however, cost up to $84,000 in the United States, which likely will not be sustainable, even though the United States has a strong health care infrastructure and relatively low HCV prevalence. 46 Gilead has agreed to reduce pricing for sofosbuvir in selected high-incidence, low-resource countries to make this drug more accessible. 46
Once treatment is initiated, quantitative HCV RNA viral load assays are performed periodically to monitor treatment, based on virological responses at various milestones ( Table 4 ). The first benchmark, the rapid virological response (RVR), helps physicians assess the appropriate treatment duration based on genotype. 47 Next, early virological response (EVR) is used to determine if the patient is a nonresponder, which if found would trigger a change in therapeutic regimen. The end-of-treatment response (ETR) is an important predictor of achieving future sustained virological response (SVR), which is used as a surrogate endpoint to determine disease remission or cure. 48
Virological Response Classification to Treatment. 11

User Needs and Requirements
The clinical algorithm for HCV diagnosis, staging, treatment selection, and monitoring shown in Figure 2 represents the standard of care in high-resource areas with well-developed laboratory infrastructure. The highest incidence and prevalence of HCV infection, however, occur in areas in the Middle East, Southeast Asia, and Northern Africa, 10 which often have limited resources and lack both suitable infrastructure and trained personnel for complex diagnostic testing methods. Therefore, to address these regional problems, a suitable test cannot require expensive consumables and large, complex, expensive laboratory equipment. Noninstrumented systems that do not require power are ideal. Minimally instrumented systems should be able to operate on battery power, because most low-resource areas lack uninterrupted power. Furthermore, reagents must have a long shelf life at elevated temperatures, due to the absence of cold chain procurement and refrigerated reagent storage. Most importantly, the test must be “so simple and accurate as to render the likelihood of erroneous results by the user negligible” to be effectively used in near-patient settings that lack trained personnel. 51 In the United States, this stipulation applies to clinical diagnostics categorized as waived under the Clinical Laboratory Improvement Amendments (CLIA). Most professional point-of-care settings, such as physician office labs and small health clinics, can perform only CLIA-waived tests.
Technical Product Analysis
Multiple in vitro diagnostic testing methods are used in HCV diagnosis, staging, treatment selection, and treatment monitoring ( Fig. 2 ). Serological assays are used for initial diagnosis by detecting anti-HCV antibodies after seroconversion. Other immunoassays and clinical chemistry assays are used to detect various biomarkers for disease diagnosis and staging ( Table 1 ). Nucleic acid testing is used to detect the viral RNA, quantify the viral load, and genotype the virus.
Serological Assays for HCV Detection
Enzyme Immunoassays
The main screening assay used to detect antibodies against HCV epitopes in plasma or serum is the EIA. 52 Although EIAs are relatively easy to use and well adapted to large-volume testing, earlier versions often had low sensitivities (i.e., the true-positive rate), meaning that not all HCV-infected patients were diagnosed with EIA screening tests.3,53,54 Furthermore, serological diagnosis requires seroconversion following acute infection. Three generations of EIAs, each with improved sensitivity, have been developed to overcome this challenge. The first-generation EIAs used viral lysate as capture antigen, whereas second-generation assays contained a recombinant HCV protein or synthetic peptide as capture antigen, such as from the virus’ NS4 gene. 53 Third- and fourth-generation EIAs usually contain multiple recombinant antigens from the HCV core; NS3; NS5; the NS4 antigen, 55 which provides higher sensitivity; and antigens recognized by immunoglobulin G (IgG) and IgM antibodies, which can produce an earlier positive response after infection. 56
Anti-HCV Serology Assays Conducted on Large Immunoanalyzers
All large core labs have immunoanalyzers with medium- to high-throughput testing capabilities, and almost every major in vitro diagnostic (IVD) company markets immunoanalyzers with HCV serology tests on the menu ( Table 5 ). Examples include the ARCHITECT (Abbott Diagnostics, Abbott Park, IL), UniCel DxI 800 (Beckman Coulter, Pasadena, CA), Cobas 4000 (Roche, Indianapolis, IN), Advia Centaur (Siemens, Berlin, Germany), and VITROS immunoanalyzers (Ortho Clinical Diagnostics, Rochester, NY).57–59 These immunoanalyzers are fully automated and optimized for efficiency and throughput. Most of these platforms use a chemiluminescent readout, which provides higher sensitivity than other detection approaches. For these HCV serology assays, capture antigens are typically immobilized on magnetic beads. An exception is the Ortho VITROS immunoanalyzer, which uses coated wells. 12 The general assay principle is depicted in Figure 3 . 60
HCV Serology Assays Performed on Large Automated Immunoanalyzers.

HCV, hepatitis C virus.
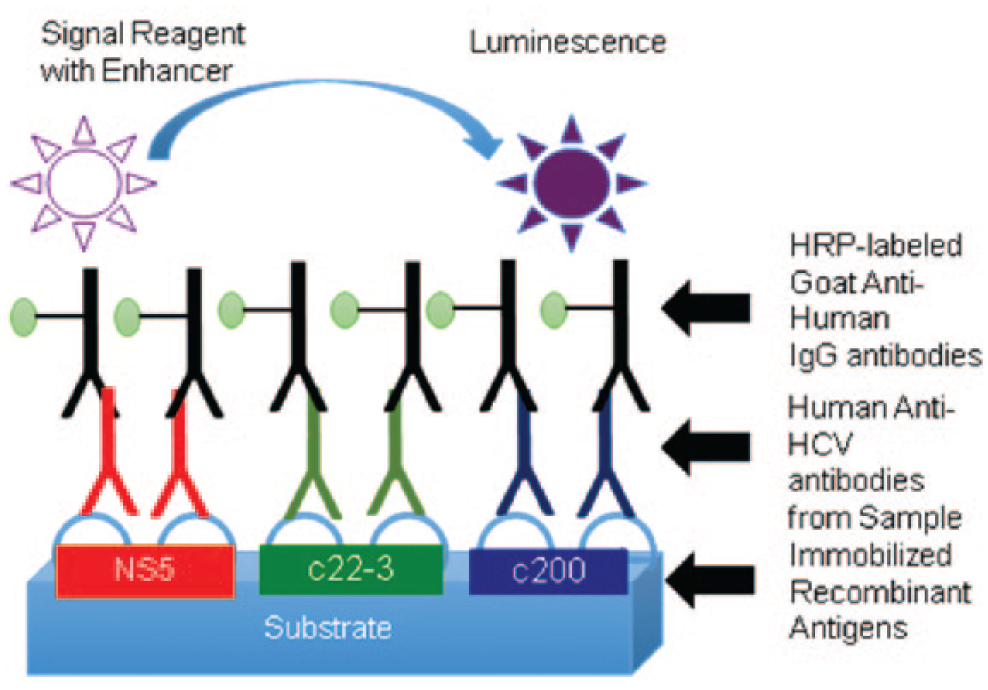
In the VITROS anti–hepatitis C virus (HCV) assay procedure, the anti-HCV antibody from the sample binds to three recombinant HCV antigens: c22-3 (a protein encoded by the core HCV genome), c200 [a protein encoded by the nonstructured protein 3 (NS3) and NS4 regions of the HCV genome], and NS5.60,61 The addition of a detection antibody [conjugated to horseradish peroxidase (HRP)] results in a luminogenic substrate being converted to a luminescent product. 62
In limited-resource settings, serology enzyme-linked immunosorbent assays (ELISAs) can be run without large immunoanalyzers in the standard plate format (e.g., using kits such as the HCV Ab ELISA kit by Diagnostic Automation/Cortez Diagnostics, Calabasas, CA). 66 These methods are, however, cumbersome and error prone. Other serology assay formats that do not require technically skilled personnel and suitable lab infrastructure are therefore preferable for low-resource settings.
Rapid Diagnostic Tests (RDTs)
RDTs that use a lateral flow immunoassay enable detection of anti-HCV antibodies for initial HCV diagnosis. As opposed to EIAs, these RDTs do not require complex instruments or skilled technical staff, can deliver results in 20 minutes, and therefore can be used in high-burden, low-resource areas.61,67 Currently, several RDTs are available on the market for HCV serology testing, including the OraQuick HCV Rapid Antibody Test (OraSure Technologies, Bethlehem, PA), the Combiquic HIV/HCV test (QualPro Diagnostics, Goa, India), 68 the Chembio DPP HCV test (Chembio Diagnostic Systems, Medford, NY), the Multiplo Rapid HIV/HCV Antibody Test (MedMira Laboratories, Halifax, Canada), ImmunoFlow (Core Diagnostics, Birmingham, UK), 69 and BioRapid HCV (BioRapid, Barcelona, Spain). 70 Orasure and Chembio currently are the market leaders in this area. As an example, the OraQuick HCV Rapid Antibody Test is described here in more detail.
OraQuick HCV Rapid Antibody Test
The OraQuick test ( Fig. 4 ) is a CE-IVD-marked, FDA-approved, and CLIA-waived rapid HCV screening assay that can be used in professional point-of-care environments, such as a doctor’s office or health care clinic. The test has been FDA approved for use with venipuncture and finger-stick whole-blood samples, 71 but it can also detect anti-HCV antibodies from oral fluid samples. 72
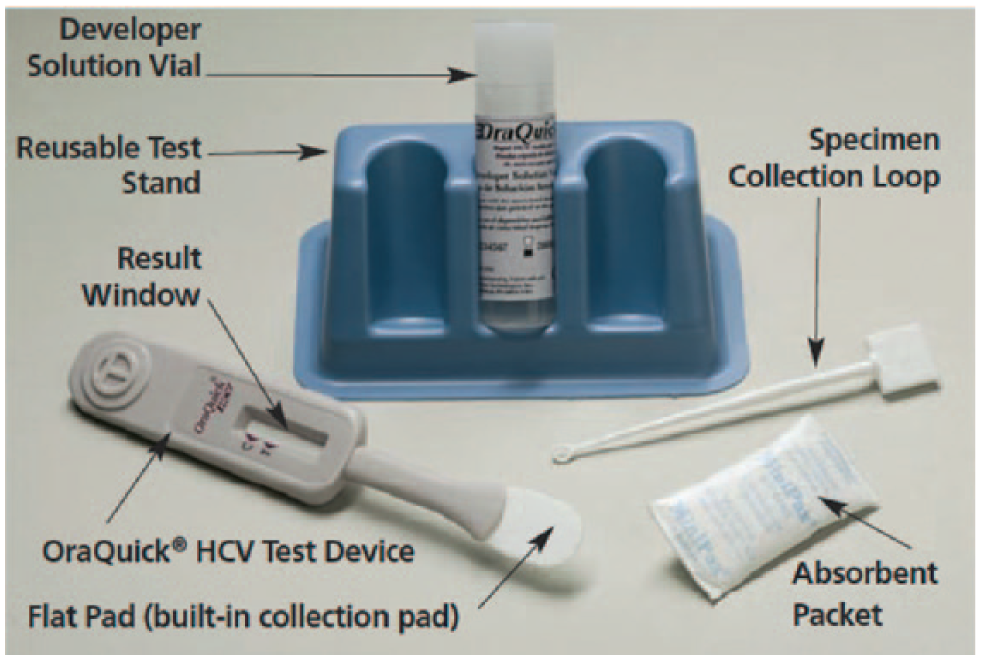
The OraQuick Test includes the single-use test device, a developer solution vial, a reusable test stand, specimen collection loops, and an absorbent packet. 73
The sample is mixed with the developer solution and then applied to the sample pad, on which it reconstitutes protein A–conjugated gold colloids deposited on the conjugate pad of the lateral flow strip ( Fig. 5 ). This protein A conjugate captures any antibodies present in the sample, which, for HCV-infected individuals after seroconversion, includes anti-HCV antibodies. The sample then migrates along the nitrocellulose membrane. If anti-HCV antibodies have been captured on the colloid, these colloids will bind to the recombinant antigens (from the core NS3 and NS4 regions of the HCV genome) that are immobilized on the test line, which then attains a red color. 67 Goat antihuman IgG antibodies immobilized on the control line recognize the human antibodies captured on the gold colloids. Therefore, the control line should always attain a red color.
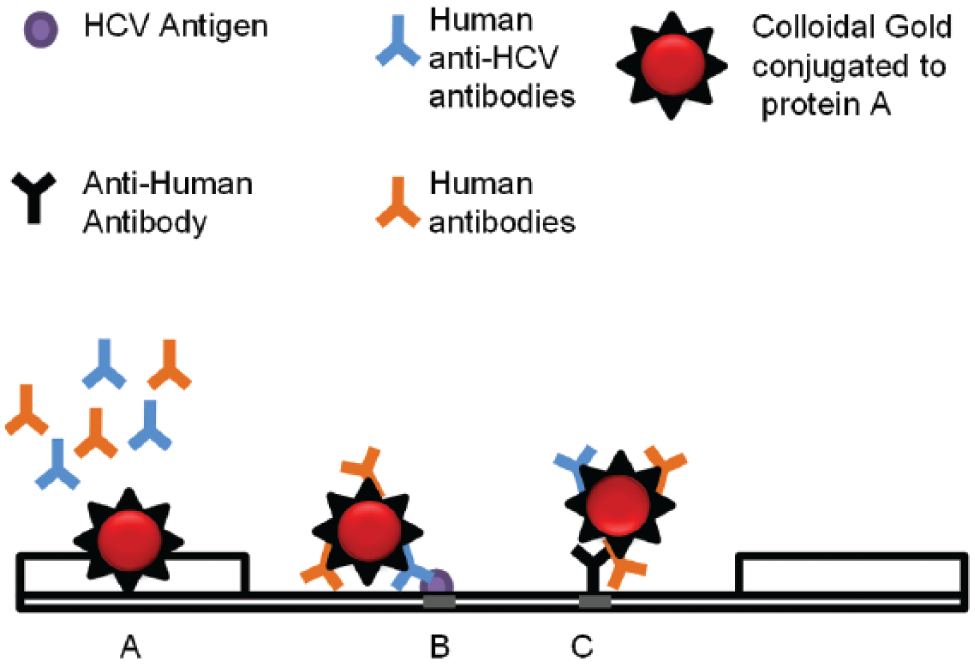
OraQuick HCV assay principle: (
The testing procedure is relatively simple: The specimen loop ( Fig. 4 ) is used to take up a certain amount of whole blood from a finger-stick or venipuncture sample. The loop is then used to transfer the sample into the developer solution vial containing a buffered saline solution. Next, the lateral flow strip device is inserted into the vial, which initiates the flow of sample and buffer through the device.71,73 A reactive, HCV-positive specimen generates red-colored test and control lines, whereas a nonreactive, HCV-negative specimen generates only a colored control line. If the control line is blank, then the test is invalid.
This OraQuick HCV assay has a sensitivity of 99.3% and specificity of 99.5% when testing from whole blood. 67 Only 5 µL of blood is required, which can be readily obtained via a finger prick. 72 Using oral fluid, the clinical sensitivity and specificity are 97.8% and 100%, respectively. 72 Testing based on oral fluid is less invasive, and medical personnel are protected from biohazards. The lower sensitivity, however, poses challenges in low-prevalence settings. In either case, the turnaround time is only 20 minutes, meaning that results can be quickly and easily delivered to the patients during their first visit. 72 The OraQuick assay therefore provides more accessibility and flexibility in low-resource, high-burden settings.61,67,72
Clinical Chemistry Assays for Assessing Liver Damage
Biomarkers for liver disease diagnosis and staging ( Table 1 ) are typically assayed via coupled enzyme assays or homogeneous immunoassays that can be performed on standard clinical chemistry analyzers. Analogous to the previously discussed immunoanalyzers, virtually all large core labs have clinical chemistry analyzers with medium- to high-throughput capabilities, with tests assaying most of the biomarkers listed in Table 1 on their standard menu. In many cases, clinical chemistry analyzers and immunoanalyzers are combined into integrated systems, such as the VITROS 5600 system by Ortho Clinical Diagnostics ( Fig. 6 ).

(
Two key biomarkers used for assessing and monitoring liver damage are aspartate aminotransferase (AST) and ALT. These biomarker levels are used in multiple combinations to identify and stage the disease in HCV patients. Two key scoring methods are AST-to-Platelet Ratio Index (APRI) and Fibrosis 4 (FIB4) scores, which are calculations based on platelet count and age. APRI is the normalized AST–platelet ratio, whereas FIB4 takes into account age, and AST and ALT values. 74 Serum levels of these biomarkers can be quantified via relatively simple coupled enzyme assays. In most systems, these reactions are implemented in a simple “mix-and-measure” format, consisting of one liquid ALT or AST reagent that contains the required substrates, enzymes, and buffer components. For example, in the Beckman Coulter Synchron AST assay, 75 a liquid AST reagent is added to the sample, and the concentration of AST is determined based on the progression of a coupled enzymatic reaction, which leads to oxidation of nicotinamide adenine dinucleotide (NADH) to NAD+, monitored via the decrease in UV absorbance at 340 nm.
However, coupled enzyme assays can also be implemented in a dry-reagent format, such as the VITROS ALT or AST slides (Ortho Clinical Diagnostics), conducted on the VITROS Chemistry Analyzers or on the VITROS 5600 Integrated System ( Fig. 6A ). Such dry-reagent formats are more compact and stable than methods based on liquid reagents. The VITROS dry-reagent slides consist of a polyester support coated with multiple layers containing the necessary reagents ( Fig. 6B ). 76 A serum or plasma sample is added to the slide, then evenly spreads throughout the multiple sublayers, thereby reconstituting the dry reagents, which initiates the reaction. Compared to liquid-reagent formats, the dry-reagent slides are less sensitive to variability in sample volume, require less frequent calibration, and have a longer shelf life when stored frozen. However, both liquid and dry slide formats require refrigerated on-analyzer reagents storage, and they remain stable on the analyzer for only a few weeks to a month.
For the VITROS ALT slide, 76 NADH is oxidized to NAD+ in the indicator reaction, and the progression of the reaction is measured through reflectance spectrophotometry in the UV range at 340 nm. In contrast, the readout of the AST slide uses a different indicator reaction, 77 generating a colored product that can be detected via reflectance readings in the visible range, at 670 nm. The lower variability in the visible range translates to higher precision, especially at lower analyte concentrations.
Such ALT and AST assays are performed routinely in developed nations with suitable laboratory infrastructure, but they are more difficult to implement in high-burden, low-resource settings. To address this unmet need, and to make ALT and AST assays suitable for point-of-care settings, a new, low-cost, paper-based, multiplexed microfluidic assay has been developed by Diagnostics for All (Cambridge, MA). This dry-reagent system is semiquantitative and involves instrument-free visual interpretation of the signal, in contrast to the VITROS dry slide ALT and AST assays, which are quantitative but require a reader to measure the signal in the near-UV or visible range. In addition, the Diagnostics for All system uses more sophisticated liquid handling in the slide, facilitated by hydrophilic channels and chambers that are generated via printing of hydrophobic wax as barriers onto two layers of paper that are sandwiched in between polyester laminates as support and cover ( Fig. 7 ). This design makes it possible to simultaneously obtain ALT and AST results from parallel assays using the same sample. The assay further incorporates integrated controls.
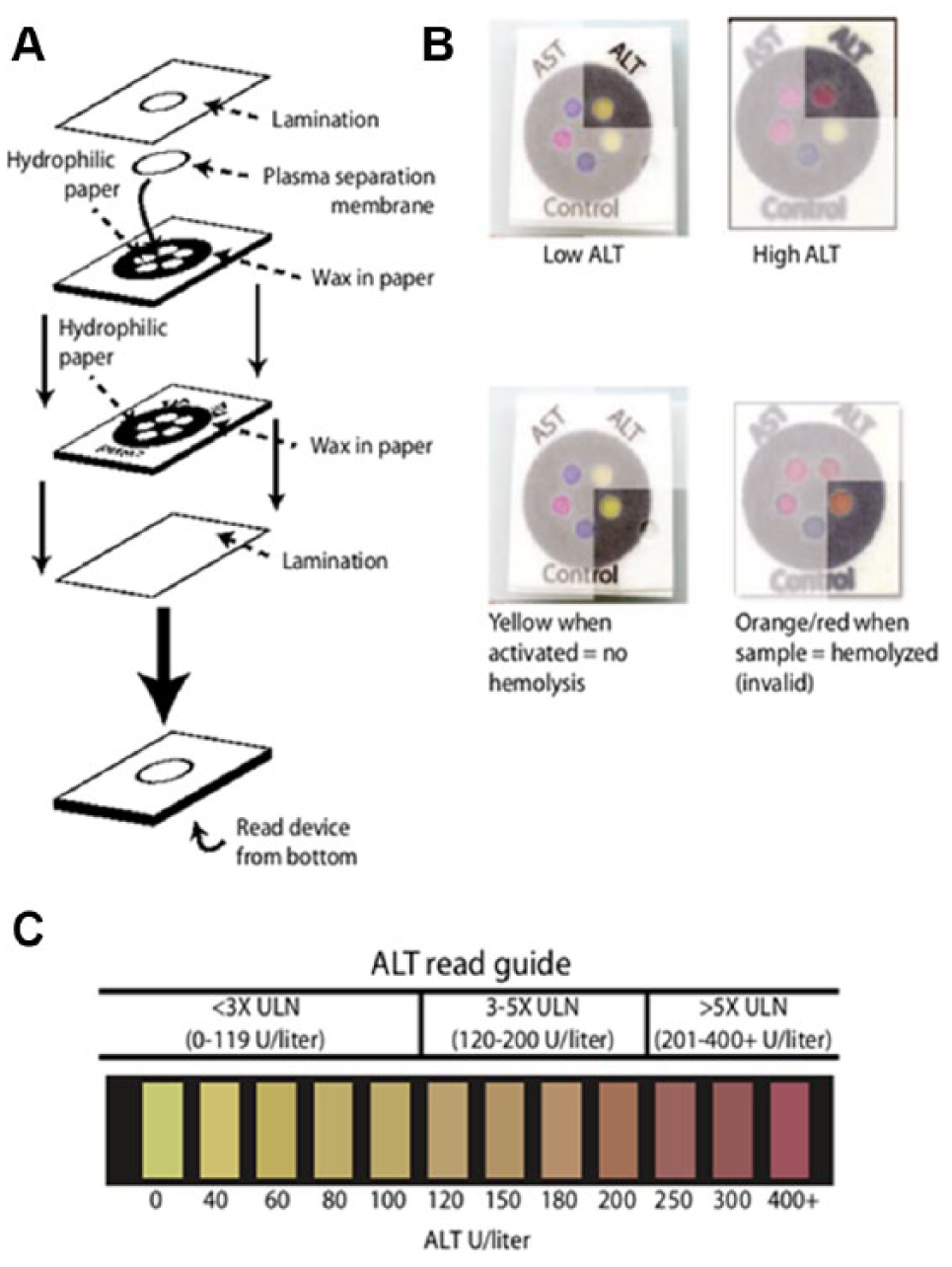
Microfluidic paper-based aspartate aminotransferase (AST) and alanine transaminase (ALT) test schematic and procedure. (
The device can use a small input sample volume (<40 μl) of whole blood obtained via a finger prick. Blood cells are filtered out via a plasma separation membrane, and then the plasma is wicked into different zones on the paper, where it reconstitutes different dry reagents to initiate the coupled enzyme assays, generating a result within 15 minutes. 78 The device can be used to differentiate three bins of high, intermediate, and low ALT or AST concentrations with greater than 90% accuracy based on visual interpretation of the result using a color gradation reference chart. However, the Diagnostics for All system has lower sensitivity and a smaller dynamic range than the VITROS slide assays. Furthermore, analyte concentration in such coupled enzyme assays is typically determined via the rate of color change. The Diagnostics for All system requires readout as an endpoint after 15 minute, which is difficult to enforce in the intended use settings. Visual interpretation further can be subjective, giving rise to interoperator variability. The rate of enzymatic conversion also depends on other parameters, such as ambient temperature, which also is highly variable in the intended use settings and cannot be controlled. Despite these shortcomings, the Diagnostics for All system has some significant advantages for the intended use settings. The device requires no external instrumentation or power source, and can be implemented at the point of care to test the patient and make treatment decisions in the same clinical encounter. Such a device can identify patients with liver damage who might benefit from antiviral treatment.
Biomarkers such as ALT and AST provide general information regarding decreased liver function, but by themselves they cannot accurately stage the severity of liver damage. Therefore, more extensive biomarker panel assays have been developed to properly stage liver disease in a minimally invasive manner. 18 Examples include the FibroTest and ActiTest (Biopredictive, Paris, France), which both measure patient serum levels of α-2-macroglobulin, haptoglobin, apolipoprotein A1, gamma-glutamyl transpeptidase (GGT), and total bilirubin. In addition, the ActiTest measures ALT as a sixth biomarker. Using an algorithm that combines the biomarker results, adjusted for the patient’s sex and age, these kits determine the fibrotic state of the liver as well as necroinflammatory activity.79,80 Compared to METAVIR scoring of liver biopsy samples, the FibroTest and ActiTest enabled differentiation between fibrosis stages F0–F2 versus F3–F4 with a receiver operating characteristic area under the curve (ROC AUC) value of 0.8. 81 At the cutoff providing the highest Youden index, the sensitivity and specificity were 76% and 70%, respectively, 81 which is relatively moderate. However, another study has demonstrated that discordant results between FibroTest–ActiTest and liver biopsy are predominantly attributable to false negatives for liver biopsy, due to a small biopsy specimen or steatosis (i.e., excessive retention of lipids in the liver). 82 This study further suggested a diagnostic algorithm, wherein liver biopsy is recommended as a confirmatory test only if the FibroTest and ActiTest do not produce coherent results. Therefore, such biomarker panel assays can complement ultrasound-based transient elastography, 18 and can reduce the need for highly invasive and expensive histopathological liver disease staging from a biopsy sample.
The FibroTest and ActiTest panels are offered by Bio-Predictive (Paris, France) as lab-developed tests (LDTs). Once collected at point-of-care settings, blood samples are shipped to BioPredictive’s central lab in which analysis is performed. These multivariate assays require algorithms to convert biomarker levels to a final liver disease score that measures fibrosis or scarring similar to a METAVIR score. The cost of the FibroTest was listed as $215 in 2009, which is more expensive than ultrasound transient elastography (listed as $131 in 2009), but much less expensive than the more invasive liver biopsy, which is priced at $1255.83,84 Liver disease staging via the FibroTest biomarker panel was found to be more cost-effective compared to liver biopsy in settings in which liver disease staging is required prior to anti-HCV treatment. 85
Molecular Assays for HCV Detection, Quantification, and Genotyping
Qualitative nucleic acid amplification tests (NAATs) are used to confirm the diagnosis of HCV infection, and on a larger scale they play a critical role in blood bank screening. Quantitative NAATs are used for monitoring viral load. Furthermore, HCV genotyping assays are used to guide treatment selection. All of these methods use nucleic acid amplification, which provides high sensitivity and specificity. 11 Most current assays and systems on the market ( Table 6 ) are targeted at high-complexity central laboratories.
HCV Molecular Diagnostic Tests for Use in Central Labs.
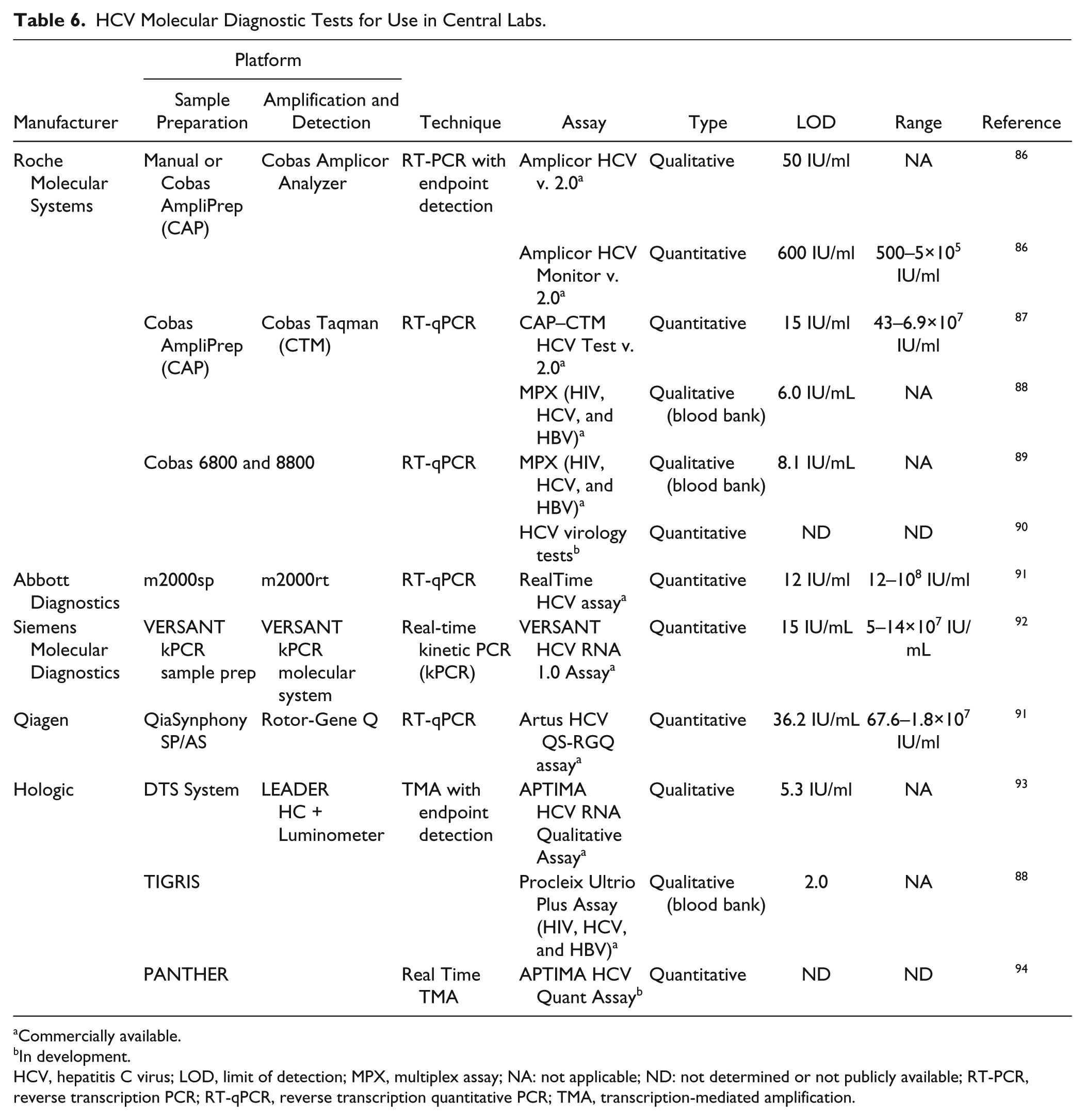
Commercially available.
In development.
HCV, hepatitis C virus; LOD, limit of detection; MPX, multiplex assay; NA: not applicable; ND: not determined or not publicly available; RT-PCR, reverse transcription PCR; RT-qPCR, reverse transcription quantitative PCR; TMA, transcription-mediated amplification.
The following sections discuss the technical principle behind several leading molecular diagnostic systems ( Table 6 ) in more detail, broken down into the three key processes required for nucleic acid testing, which are sample preparation, amplification, and detection. Much progress has been made to integrate and automate the entire workflow, improve ease of use, standardize processes, increase throughput, and mitigate the risk of amplicon carryover contamination.
Sample Preparation
Nucleic acid sample preparation, in this case HCV RNA extraction from plasma, is often the bottleneck in NAATs because it involves cumbersome and lengthy protocols. Low extraction yield, failure to remove inhibitors, and RNA degradation can compromise test results. Sample preparation in most currently used systems, including the Cobas AmpliPrep (Roche), m2000 (Abbott Molecular, Abbott Park, IL), and VERSANT kinetic Polymerase Chain Reaction (kPCR) sample prep system (Siemens), involves automated solid phase extraction, wherein nucleic acids reversibly adsorb to silica-coated magnetic beads in the presence of chaotropic salts and organic solvents. After multiple wash steps to remove sample matrix and other inhibitors, purified nucleic acids are eluted into a low ionic strength buffer suitable for subsequent polymerase amplification. As alternative to silica-based solid phase extraction, the Direct Tube Sampling (DST) System, TIGRIS and PANTHER platforms (Hologic, Bedford, MA), uses probe-based capture for RNA extraction. Capture oligonucleotides hybridize to the HCV target RNA through a complementary region at the 5′ end. This capture probe also contains a 3′ poly-A tail, through which the probe–target duplex is then captured by magnetic beads conjugated to poly-T oligonucleotides. As before, sample matrix and other inhibitors are removed via magnetic separation and washing.
In terms of process automation, these systems typically entail a liquid handling platform, onto which the operator manually loads the sample input tubes, sample preparation reagents, and other consumables such as disposable tips, custom reaction containers, and final output tubes for extracted nucleic acids. The instrument then performs reagent addition, mixing, magnetic separation, washing, and heating steps. Automation challenges include the need to go from large initial volumes (sample plus lysis buffer, and wash buffer) to small final volumes (elution buffer and eluate) with minimal sample loss. Effective mixing is needed to facilitate RNA binding to and release from the magnetic beads.
However, the level of integration varies among these platforms. The Siemens VERSANT kPCR Sample Prep system, Abbott m2000sp platform, and Qiagen Qiasymphony SP/AS (Qiagen, Venlo, the Netherlands) are stand-alone liquid handling robots that automate nucleic acid extraction and assay setup, wherein final eluates are combined with master-mix reagents. However, tubes containing the final master-mix have to be manually transferred to the amplification and detection instrument. The Cobas AmpliPrep (CAP) sample preparation instrument can be docked with the Cobas Taqman (CTM) system for amplification and detection, with automated transfer of tubes containing the final master-mix via a conveyor belt. The Hologic TIGRIS was the first NAAT system to enable nucleic acid sample preparation, amplification, and detection in an integrated and fully automated manner in one instrument. This high-throughput system automates the Procleix Ultrio Assay for multiplexed, qualitative detection of HIV, HCV, and hepatitis B virus (HBV) in blood products (marketed by Novartis Molecular Diagnostics, Basel, Switzerland). The TIGRIS enables DTS from primary sample tubes via pierce-able caps. The sample is transferred into a reagent tube within a multitube unit (MTU), where it is mixed with lysis buffer and other sample preparation reagents. The MTU is moved via a circular robotic arm to various stations that enable mixing, incubation to enable lysis and target capture, magnetic separation, and washing. Hologic also markets the APTIMA HCV RNA qualitative assay for initial diagnosis of HCV infection and to determine treatment success. However, this APTIMA assay is not approved for implementation on the TIGRIS platform. Instead, sample preparation based on the same technology as implemented in the TIGRIS can be automated on a separate instrument called the DTS System. Hologic is currently developing the APTIMA HCV Quant Assay for quantitative HCV viral load monitoring, to be implemented on the PANTHER instrument, a more compact and refined version of the TIGRIS. 95 Roche Molecular Systems recently received CE-IVD approval for the Cobas 6800 and Cobas 8800, two high-throughput, fully integrated nucleic acid testing systems that automate sample preparation, amplification, and detection in one instrument. 90 The initial assay menu on launch focused on pathogen detection for blood bank screening, but an HCV viral load assay is in development and expected to be launched in the near future.
Amplification and Detection
After RNA extraction, conserved sequences in the HCV genome are amplified and then detected. In most cases, this entails polymerase-based target amplification via PCR or isothermal methods, coupled with endpoint or real-time detection formats. Methods based on signal amplification, such as the VERSANT bDNA assays, 96 are being phased out. Because HCV is an RNA virus, PCR amplification has to be preceded by reverse transcription (i.e., RT-PCR). RT-PCR can be coupled with endpoint detection, as in the Cobas Amplicor analyzer, which, however, requires additional liquid handling steps after amplification, introducing the risk of carryover contamination. In contrast, assays based on quantitative real-time RT-PCR (RT-qPCR) perform amplification and detection simultaneously in a closed tube format, which mitigates these concerns. Furthermore, RT-qPCR is more accurate and provides a larger dynamic range than endpoint formats. For this reason, Roche is phasing out the AMPLICOR assays in favor of the Cobas TaqMan RT-qPCR HCV Assay, for which version 2 was released in 2013. 97 The assay uses primers specific to conserved sequences within the 5′-untranslated region (5′ UTR) of the HCV genome. Real-time detection is facilitated through TaqMan probes labeled with a fluorophore and quencher, 98 which are cleaved as the reaction progresses, based on the intrinsic 5′-exonuclease activity of the polymerase used in this assay. 97 The risk of false positives due to carryover contamination is reduced by including the enzyme uracil N-glycosylase (UNG), also called AmpErase, in the master-mix. 99 The risk for false negatives due to failed RNA extraction or presence of polymerase inhibitors in the reaction is mitigated by including a full-process internal amplification control (IAC), which consists of an armored RNA containing a sequence that is amplified by the same primers as the HCV target. However, the IAC contains a different internal sequence, which is detected by a different Taqman probe with a different fluorophore, in a separate channel of the real-time fluorometer.
Other RT-qPCR assays currently on the market, such as the Abbott RealTime and Siemens VERSANT kPCR HCV viral load assays, use a similar general format, with some variations in assay implementation and probe chemistry. Both assays likewise target the 5′ UTR region of the HCV genome. The Siemens kPCR assay also uses Taqman probes, whereas the Abbott RealTime HCV assay uses hybridization probes labeled with a fluorophore and quencher that are not cleaved during the reaction. 100 The probe is quenched when present as a single-strand random coil in solution, but it recovers fluorescence when bound to the amplicon. The Abbott RealTime and Siemens VERSANT kPCR HCV viral load assays also incorporate UNG to mitigate carryover contamination, and they include a full-process IAC in the form of armored RNA. However, both assays use a noncompetitive format, wherein the IAC target is amplified using different primers than the HCV target, in contrast to the competitive format used in the Roche Cobas TaqMan assay.
Isothermal methods for detection and quantification of HCV viral RNA include transcription-mediated amplification (TMA), 101 nucleic acid sequence-based amplification (NASBA), 102 and loop-mediated isothermal amplification (LAMP).103,104 TMA-based HCV assays are commercialized by Hologic. TMA and NASBA use RNA polymerase-mediated amplification, in contrast to PCR and LAMP. In the TMA reaction scheme, 101 the viral RNA is reverse transcribed using a primer with 5′ overlap containing the promoter sequence of the bacteriophage T7 RNA polymerase. The RNA strand in the so-generated RNA–DNA duplex is degraded via RNAse H, producing single-stranded (ss) cDNA. Via a second primer and using the DNA polymerase activity of the reverse transcriptase, ss cDNA is then converted into a double-stranded (ds) DNA template with the T7 promoter region at the upstream end. The T7 RNA polymerase recognizes the ds promoter region, and isothermally generates hundreds to thousands of amplicons via in vitro transcription. The amplicons feed back into the cycle, generating new dsDNA templates, causing exponential amplification. The reverse transcriptase used in TMA contains endogenous RNAse H activity, whereas a separate RNAse H enzyme has to be included in NASBA. Other than that, the two reactions are identical. The Hologic TMA assays use the hybridization protection assay (HPA) to detect the RNA amplicons. 105 A probe sequence labeled with an internal acridinium ester hybridizes to the target RNA amplicon, which protects the acridinium from hydrolysis. Unhybridized probes are then hydrolyzed, removing background signal. During the subsequent detection step, the hybridized probe is dissociated from the target and hydrolyzed, generating a chemiluminescent signal. By using probes with different hydrolysis kinetics (called flashers versus glowers), an internal amplification control can be incorporated into the reaction. However, the HCV TMA assays currently marketed by Hologic with HPA-based endpoint detection are not quantitative and cannot be used for viral load monitoring. Currently, a quantitative real-time HCV TMA assay is in development for the new PANTHER platform.
RT-LAMP is another isothermal amplification method that can enable qualitative HCV detection.103,104,106 RT-LAMP uses between four and seven primers to recognize multiple complementary regions within the 5′ UTR of the viral genome, generating high-molecular-weight concatenated amplicons. These amplicons can be detected via a visual turbidimetric readout with fluorescence enhancement. 107 Such LAMP assays do not require expensive thermocyclers and detection instruments, which is an advantage for low-resource settings. However, sample preparation and reaction setup typically has to be performed manually, which impedes implementation in settings with minimal laboratory infrastructure and with users of low skill level.
Method Comparison
Patient treatment decisions depend on the accurate determination of their HCV viral load, which can range from >1e7 IU/mL plasma at baseline, prior to treatment initiation, to <10 IU/mL plasma, the viral load threshold required to be considered undetectable.49,50 Therefore, HCV viral load assays need to have a low limit of detection (LOD) and large dynamic range. Most but not all currently available HCV viral load assays are able to cover this large clinically relevant range ( Table 6 ). To ensure accurate results, HCV viral load assays are calibrated against common reference standards provided by the World Health Organization (WHO). 108 However, the accuracy of viral load quantification also depends on the genotype. Although good concordance was observed between the Abbot RealTime and Roche Cobas TaqMan HCV viral load assays for genotypes 1–4, 100 the Qiagen Artus assay overestimated the concentration of genotypes 4 and 5 compared to the Abbott RealTime assay. 91 In contrast, the Siemens VERSANT kPCR assay significantly underestimated the HCV viral load in samples of subtypes 2a, 3a, and 4a. 109 Currently, the Abbott RealTime HCV assay and the Roche Cobas TaqMan HCV assay appear to be the gold standards for HCV RNA quantification. Both assays were highly accurate at predicting an SVR for HCV patients at week 12. 100
HCV Genotyping Assays
HCV genotyping is essential to inform appropriate treatment selection. 110 The predominant HCV genotype infecting a patient can be determined by analyzing genotype-specific sequences of the viral genome, for example within the 5′ UTR, core, E1, or nonstructural region 5b (NS5B). 111 HCV genotyping methods include direct sequencing, RT-qPCR, and hybridization-based line probe assays. 112
Direct Sequencing
The current gold standard for HCV genotyping is direct sequencing of specific regions within the viral genome, followed by alignment to reference sequences and phylogenic analysis. 111 These methods are used in epidemiologic studies 113 and have been implemented in the form of LDTs, which are offered for clinical diagnostic purposes by reference laboratories. An example is the HCV DupliType assay offered by Quest Diagnostics (New York, NY). 114 However, these LDTs can be offered by only the CLIA-certified, high-complexity reference laboratories that developed the tests. Siemens has commercialized the TRUGENE HCV Genotyping Assay,115,116 wherein a 244 base pair fragment of the 5′ UTR of the HCV viral genome is amplified via RT-PCR, then subjected to bidirectional CLIP (cross-linking immunoprecipitation) sequencing, on the compact and integrated OpenGene DNA sequencing system (Siemens). The TRUGENE HCV Genotyping Assay is commercialized for research use only, although the related TRUGENE HIV Genotyping Assay has been cleared for IVD use by the FDA.
Commercially available FDA-approved and/or CE-IVD-marked tests for HCV genotyping include the RealTime HCV Genotype II assay and the VERSANT HCV Genotype 2.0, a hybridization-based line probe assay.
RT-qPCR
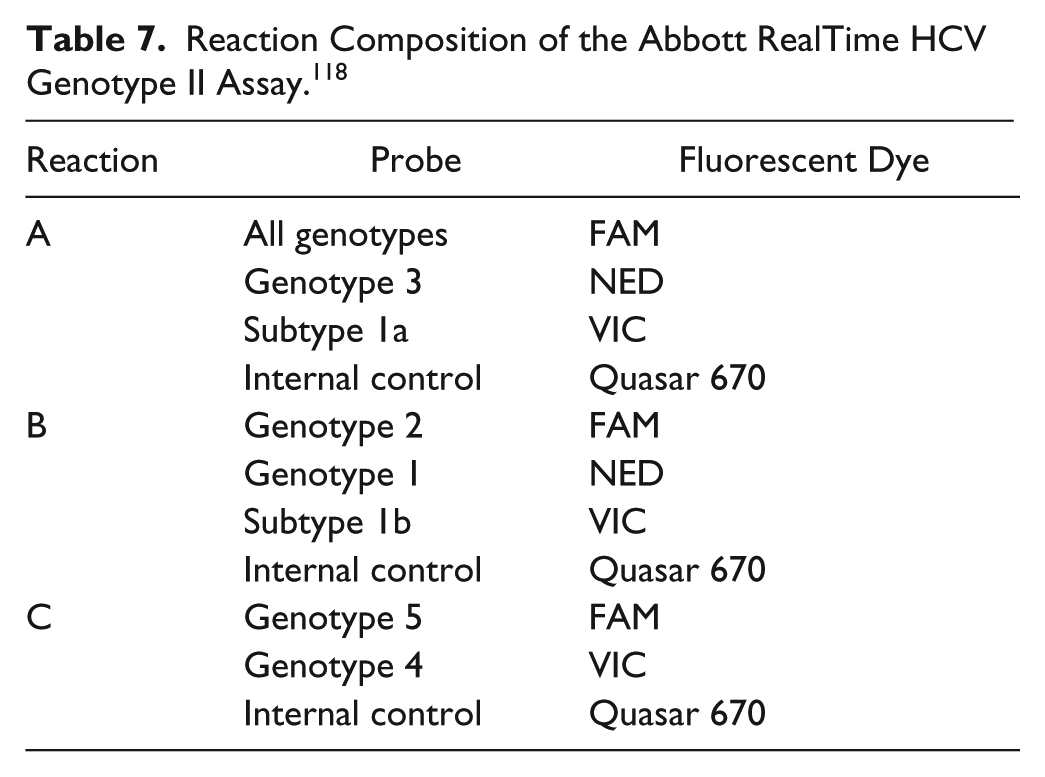
The RealTime HCV Genotype II assay (Abbott) has been CE-IVD marked and FDA approved for clinical diagnostic use. This system uses RT-qPCR with Taqman probes to detect and differentiate the most prominent HCV genotypes. 117 Each sample is analyzed via three separate RT-qPCR reactions ( Table 7 ), containing primers and genotype-specific and internal control–specific probes conjugated to different fluorophores, to enable multiplexed detection. The assay entails four different primer sets: One primer set amplifies a sequence in the 5′ UTR present in all genotypes; the second and third primer sets amplify sequences in the NS5B regions present in genotypes 1a and 1b, respectively; and the fourth primer set amplifies an IAC added to the sample upstream of sample preparation in the form of an armored RNA. Sample preparation, amplification, and detection are automated on Abbott’s m2000 platform, the same system used for the RealTime HCV viral load assay.
Reaction Composition of the Abbott RealTime HCV Genotype II Assay. 118
Line Probe Assays
The VERSANT HCV Genotype 2.0 test (Siemens) and the LINEAR ARRAY Hepatitis C Virus Genotyping Test (Roche) are CE-IVD marked, and thus can be used in Europe for HCV genotyping of chronically infected patients. In the VERSANT HCV Genotype 2.0 test, 119 sequences in the 5′ UTR and core regions of the HCV genome are first amplified via RT-PCR using biotinylated primers. The resulting biotinylated amplicons are then hybridized to genotype-specific oligonucleotide probes immobilized as lines on strips of nitrocellulose membrane ( Fig. 8 ). Next, a streptavidin–alkaline phosphatase conjugate is added, which then binds to the biotinylated captured amplicons. Following a wash step, a chromogenic substrate (BCIP–nitrobluetetrazolium) is added, forming a purple–brown product that precipitates at each line where amplicons have been captured ( Fig. 9 ). The hybridization, wash, and development steps can be automated using instruments such as the AutoBlot 3000 and LiPA 48. Result interpretation is facilitated by the LiPA HCV Scan software.
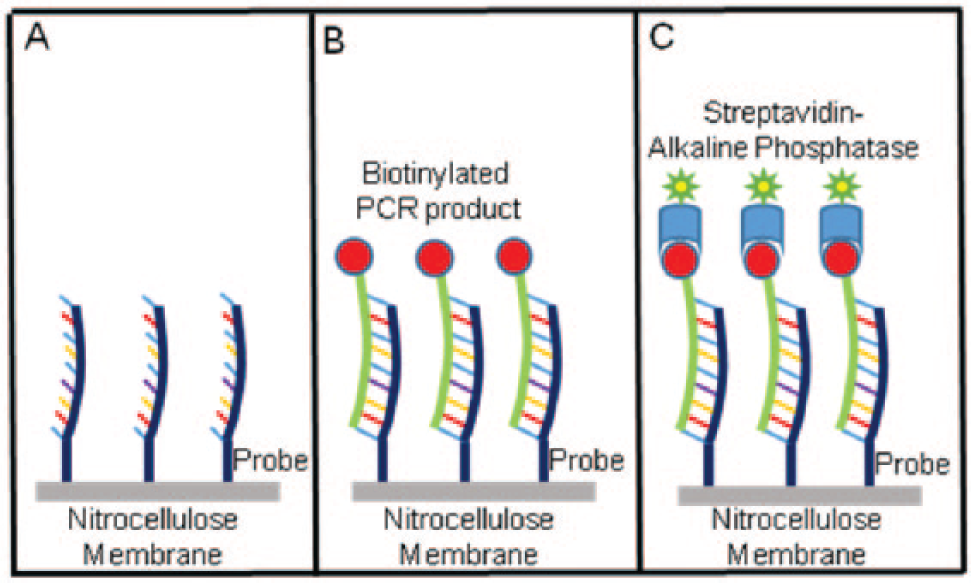
(
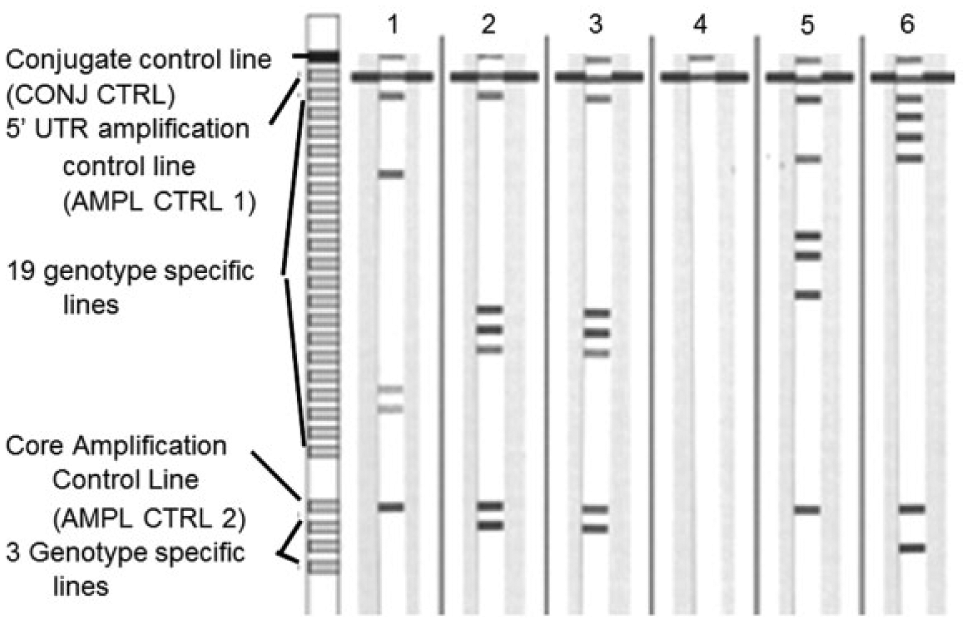
VERSANT HCV Genotype 2.0 line probe assay: strip design and sample results. Strip 1: genotype 4; Strips 2 and 3: genotype 3a; Strip 4: negative control; Strip 5: genotype 2b; Strip 6: genotype 1a. Figure from Verbeek et al. 120 with permission.
The Roche LINEAR ARRAY test uses the same line probe assay principle, but it uses HRP instead of alkaline phosphatase for signal amplification, with a substrate that gives rise to a blue precipitate. This test identifies genotype-specific sequences present in the biotinylated 5′ UTR amplicon generated by the Cobas Amplicor assay.
Method Comparison
Direct sequencing is considered the gold standard in HCV genotyping, and it is frequently used to resolve failed analysis or discordant results obtained using other methods. 121 However, direct sequencing is not frequently implemented in routine clinical laboratories, mainly due to technical challenges and a lack of FDA-cleared or CE-IVD-marked methods. Furthermore, compared to RT-qPCR and line probe assays, direct sequencing has limitations when genotyping low titer samples or mixed infections. 116 Methods that only target sequences within the 5′ UTR cannot accurately subtype genotypes 1a and 1b, or differentiate between genotypes 1 and 6.112,119 These methods include the sequencing-based Siemens TRUGENE HCV genotyping assay, the Roche Linear Array line probe assay, and an earlier version of the Siemens VERSANT line probe assay. Newer methods include additional targets, such as sequences in the NS5B or core regions, to improve overall genotyping accuracy. Line probe assays have been partially automated, but they nevertheless require multiple manual operator steps and handling of amplified master-mix, which introduce risk of carryover contamination. Of the HCV genotyping methods discussed in this article, the Abbott RealTime HCV Genotype II assay has the lowest LOD, 117 and it is the only fully automated assay, which translates into improved ease of use and reduced time to result.118,122
Discussion and Conclusions
Most of the diagnostic methods discussed herein are mature and routinely used in high-resource countries. However, these methods are typically restricted to centralized laboratories with well-developed infrastructure. Technologies suitable for low-resource settings must follow guidelines indicated by the acronym ASSURED, coined by the WHO in 2009. This acronym stands for affordable, sensitive, specific, robust and rapid, equipment free or minimally equipped, and deliverable (e.g., reagents are thermo-stable and there are no cold chain requirements). 123
In 2014, the WHO published the first guidelines for HCV screening, care, and treatment, which provides recommendations for the use of some but not all technologies discussed in this report ( Table 8 ). 74
Applicability of HCV IVD Tools for Use in High-Burden, Low-Resource Settings.
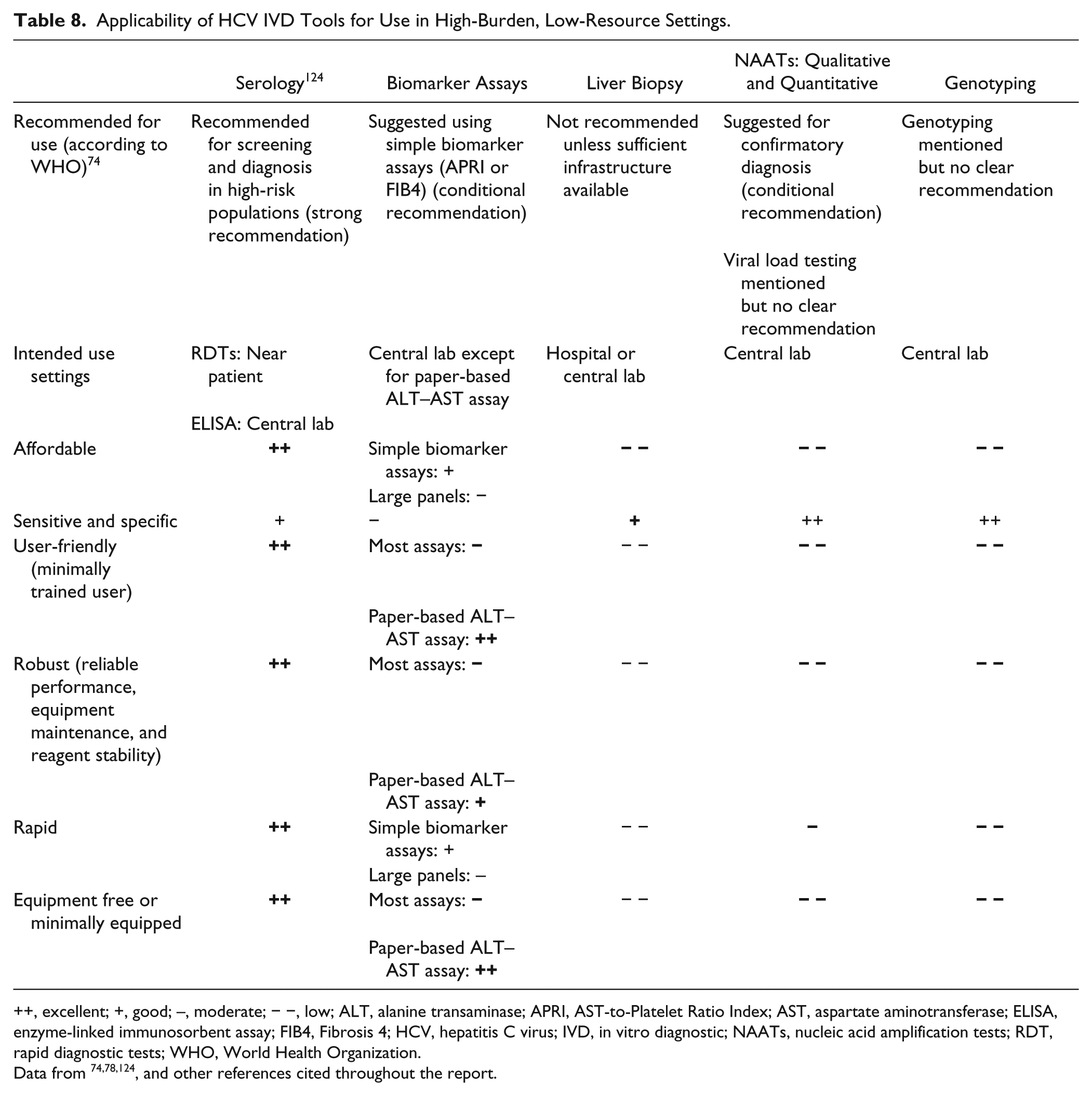
++, excellent; +, good; –, moderate; − −, low; ALT, alanine transaminase; APRI, AST-to-Platelet Ratio Index; AST, aspartate aminotransferase; ELISA, enzyme-linked immunosorbent assay; FIB4, Fibrosis 4; HCV, hepatitis C virus; IVD, in vitro diagnostic; NAATs, nucleic acid amplification tests; RDT, rapid diagnostic tests; WHO, World Health Organization.
Lateral flow serological assays used for initial HCV diagnosis are the exception. These assays are mostly performed on high-risk groups, although recent changes in CDC guidelines have increased the market size for HCV screening assays in the United States. RDT lateral flow assays are most suited for initial screening and HCV diagnosis in high-risk marginalized populations or in low-resource settings, where patients have limited access to care or do not seek care, and may drop out if results are not immediately available. The only CE-IVD-marked and FDA-approved RDT on the market is the OraQuick HCV test (OraSure Technologies), which can be performed in professional point-of-care settings or at home by the patient. In the United States, the OraQuick test is reimbursed under Medicare and other health insurance plans. However, the OraQuick test is 4–12 times more expensive than other HCV RDTs that have not undergone regulatory review and approval.125,126 The cost likely will limit uptake in low-resource settings unless suitable pricing arrangements are implemented. WHO prequalification via the PQDx system is required for public sector procurement in global low-resource regions. At this point, there are no HCV RDTs on the WHO PQDx list, and only a single lab-based EIA is listed as “in progress.” 127 However, it is anticipated that this will change in the future. WHO guidelines for serological diagnosis of HCV via simple RDTs require sensitivity ≥98% and specificity ≥97% to avoid an excessive number of false positives and missed cases. The diagnostic accuracy of currently available RDTs, however, varies widely. 125 Furthermore, some lateral flow HCV assays require serum or plasma, which cannot be readily obtained from whole blood in low-resource point-of-care settings. False negatives are a major concern in patients with HCV–HIV co-infection, because immunosuppression and anti-HIV therapy can alter the accuracy of HCV screening results. 125
Various biomarker assays can be performed to assess liver function and enable disease staging, often in conjunction with ultrasound imaging, as alternatives to liver biopsy. However, currently available biomarker tests, including biomarker panels, have moderate diagnostic accuracy for liver disease staging. 18 The WHO currently recommends the APRI and FIB4 scores based on ALT and AST activities as liver biomarkers for disease staging. Larger biomarker panels such as the FibroTest and ActiTest provide increased accuracy, but are not recommended by the WHO due to cost and infrastructure limitations. 74 To enable the assessment of liver function, the company Diagnostics for All has developed a paper-based ALT and AST assay, which despite some limitations can be used in low-resource point-of-care settings. This paper-based microfluidic assay is still in development, but in principle it would be applicable in high-burden, low-resource settings because it complies with most of the ASSURED guidelines discussed in Table 8 . 78
Qualitative HCV molecular assays are used for blood bank screening and to confirm initial diagnoses. Viral load and genotype assays are used to guide HCV treatment. Both qualitative and quantitative molecular HCV assays are moving from endpoint toward real-time detection, in general in closed tube formats with other mitigation strategies to prevent amplicon carryover. Most of the HCV NAAT systems discussed herein are highly automated and are moving toward full-process integration, including sample preparation, amplification, and detection. The WHO guidelines 74 suggest the use of qualitative HCV NAATs for confirmatory diagnosis. Furthermore, the guidelines discuss the merit of HCV viral load assays and include treatment algorithms that are contingent on viral load quantification for treatment monitoring. The guidelines, however, contain no clear recommendation for implementing viral load monitoring, because the required infrastructure is not available in most high-burden, low-resource countries.
HCV genotyping currently is an important aspect in patient management, because peg-interferon-based HCV treatment approaches are dependent on the dominant genotype. HCV genotyping prior to treatment initiation is not performed in most high-burden, low-resource settings, however, due to the high cost and the lack of suitable infrastructure. 74 This results in pragmatic decision making (i.e., presumptive treatment selection based on the predominant genotype in the geographic location). Unfortunately, this can be done effectively in only a few countries such as Egypt, where a large percentage of the population is infected with a singular genotype. 74 The new pipeline of anti-HCV drugs, however, focuses on a more pan-genotypic approach, which if successful could change the diagnostic algorithm and make genotyping obsolete.
In conclusion, many well-established assays and systems for HCV diagnosis, treatment selection, and monitoring are on the market. Few technologies are available, however, that address the needs of low-resource areas with high HCV prevalence, such as Africa and Southeast Asia. HCV infects more than 200 million people globally with increasing incidence, especially in developing countries. HCV infection frequently progresses to chronic liver disease, creating a heavy economic burden on resource-poor countries and lowering patient quality of life. Appropriate technologies that enable HCV diagnosis, treatment selection, and treatment monitoring are important in stopping disease progression.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.