
Abstract
Study Design:
Systematic review.
Objectives:
Superiority claims for medical devices are commonly derived from noninferiority trials, but interpretation of such claims can be challenging. This study aimed to (a) establish the prevalence of noninferiority and superiority designs among spinal device trials, (b) assess the frequency of post hoc superiority claims from noninferiority studies, and (c) critically evaluate the risk of bias in claims that could translate to misleading conclusions.
Methods:
Study bias was assessed using the Cochrane Risk of Bias Tool. The risk of bias for the superiority claim was established based on post hoc hypothesis specification, analysis of the intention-to-treat population, post hoc modification of a priori primary outcomes, and sensitivity analyses.
Results:
Forty-one studies were identified from 1895 records. Nineteen (46%) were noninferiority trials. Fifteen more (37%) were noninferiority trials with a secondary superiority hypothesis specified a priori. Seven (17%) were superiority trials. Of the 34 noninferiority trials, 14 (41%) made superiority claims. A medium or high risk of bias was related to the superiority claim in 9 of those trials (64%), which was due to the analyzed population, lacking sensitivity analyses, claims not being robust during sensitivity analyses, post hoc hypotheses, or modified endpoints. Only 4 of the 14 (29%) noninferiority studies provided low bias in the superiority claim, compared with 3 of the 5 (60%) superiority trials.
Conclusions:
Health care decision makers should carefully evaluate the risk of bias in each superiority claim and weigh their conclusions appropriately.
Keywords
Introduction
Randomized controlled trials (RCTs) are pivotal in establishing the safety and efficacy of novel spinal devices. Spinal device trials are designed either as noninferiority (NI) or superiority trials. In NI trials, the aim is to demonstrate that an investigational device is similar to an accepted surgical procedure or device by showing that the investigational device is not worse (by a small margin). In superiority trials, the goal is to show that the investigational device is superior to a control treatment, which may be nonsurgical care or a gold standard surgical procedure. 1
In the United States, most investigational device exemption (IDE) studies of novel spinal devices are designed as NI trials because of effect size, secondary benefits, and ethical considerations. 2,3 Many of these NI trials also test for superiority of the investigational device (NI + S), since sponsors are under pressure from physicians and payers to show improvements in safety, efficacy, and cost-effectiveness. The nuances associated with post hoc tests of superiority in this setting can make interpretation of such superiority claims challenging and potentially misleading. 4 The aims of NI versus superiority trials differ substantially, so the methodology associated with design, analysis, and interpretation is also different. For example, it is conservative to analyze the intention-to-treat (ITT) population for superiority analyses, but it is not conservative for NI analyses since any confounding events will drive the result toward equivalence. 1,5 -7 Additionally, post hoc specification of hypotheses must be avoided in confirmatory trials, 4 which requires that superiority analyses are well-defined in the statistical plan a priori. Finally, it is critical to address not only the statistical superiority but also the clinical significance of the differences observed. This is particularly true when a NI margin is imposed for the primary analysis, so that interpretation can be symmetric with less potential for bias. 8
The purpose of this study was to review the literature for reports of randomized controlled trials of spinal devices from the year 2000 to present. For each report, the primary study design was classified as NI, superiority, or NI with an additional predefined superiority analysis (NI + S). For each trial, superiority claims were identified and were assessed for potential sources of bias by multiple reviewers using a standardized tool. The hypothesis was that NI trials would predominate, and that superiority claims derived from NI trials would have a greater risk of potential bias and less reliability.
Methods
Study Selection
This systematic review was performed according to the guidelines provided by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement. 9 Search criteria were developed to identify RCTs of medical devices or biologics for the spine through PubMed/MEDLINE, Embase, ClinicalTrials.gov, the World Health Organization’s International Clinical Trials Registry Platform (ICTRP), as well as the Food and Drug Administration’s (FDA) databases on premarket approvals (PMA), postapproval studies (PAS), and proceedings from FDA advisory committee meetings of the Orthopaedic and Rehabilitation Devices Panel. Search filters included information available in the English language and publication since the year 2000 to focus on more recent trends in trial design, analysis, and interpretation. Search terms and inclusion/exclusion criteria for record screening are summarized in Table 1 for the PubMed and Embase searches while further details for these and the other databases are provided in Appendix A (see Supplementary Material available in the online version of the article). Two independent researchers screened the identified records for inclusion and exclusion. The final search of each database was completed between May 15 and June 15, 2018. When relevant studies were identified through one database, the other databases were further queried to identify protocols or reports that may provide supplemental study information for data extraction. Only the primary endpoint and primary outcomes were evaluated in this review, considering those were the basis for trial design.
Search Terms and Screening Criteria Used for the PubMed and Embase Databases.

Data Extraction
Relevant data was extracted from each included study by 2 independent researchers. Discrepancies in identifying the study design were resolved through discussion and identification of additional, clarifying documentation in five cases. The data of interest for this review included the study objective, hypotheses for primary endpoints (NI or superiority), margins or effect size used in trial design, primary outcomes and endpoints, sample sizes, conclusions or claims made in the report (NI or superiority), treatment effects of superior devices, and any statistical or clinical significance considerations relating to the superiority claims. When multiple articles reported on the same study (eg, outcomes at different time points), each article was screened for the data of interest related to the a priori study design and primary endpoint.
Risk of Bias Assessment
The general risk of bias was evaluated for each study using the Cochrane Risk of Bias Tool for Randomized Controlled Trials 10 and interpreted according to the key domains described by Pavon et al 11 (Supplementary Table S1, Appendix B). The reporting of any financial disclosures, or lack thereof, was also noted but was not considered in the overall risk of bias evaluation. Additionally, the risk of bias specifically related to superiority claims was assessed. The criteria for this assessment included analyses that were not specified a priori, analysis of the ITT population, post hoc modification of primary outcomes, and any sensitivity analyses performed on the analysis population or missing value imputation (Supplementary Table S2, Appendix B). The reporting of confidence intervals for superiority claims was also noted but was not considered in the overall risk of bias for the superiority claim.
Results
Overview of Included Studies
Across all 7 databases, 1895 unique records were identified, and 41 unique studies met the inclusion/exclusion criteria (Figure 1). Among these 41 studies, the most common investigational spinal devices were cervical disc replacements (9/41; 22%), followed by interspinous/interlaminar spacers (7/41; 17%), biologics used to support spinal fusion (7/41; 17%), lumbar disc replacements (6/41; 15%), vertebroplasty materials (2/41; 5%), spinal cord stimulators (2/41; 5%), interbody fusion cages (2/41; 5%), and 1 each (2%) of a dural sealant, an adhesion barrier gel, an annular closure device, a sacroiliac joint fusion device, a dynamic posterolateral pedicle screw system, and a surgical robot used for pedicle screw placement (Table 2).
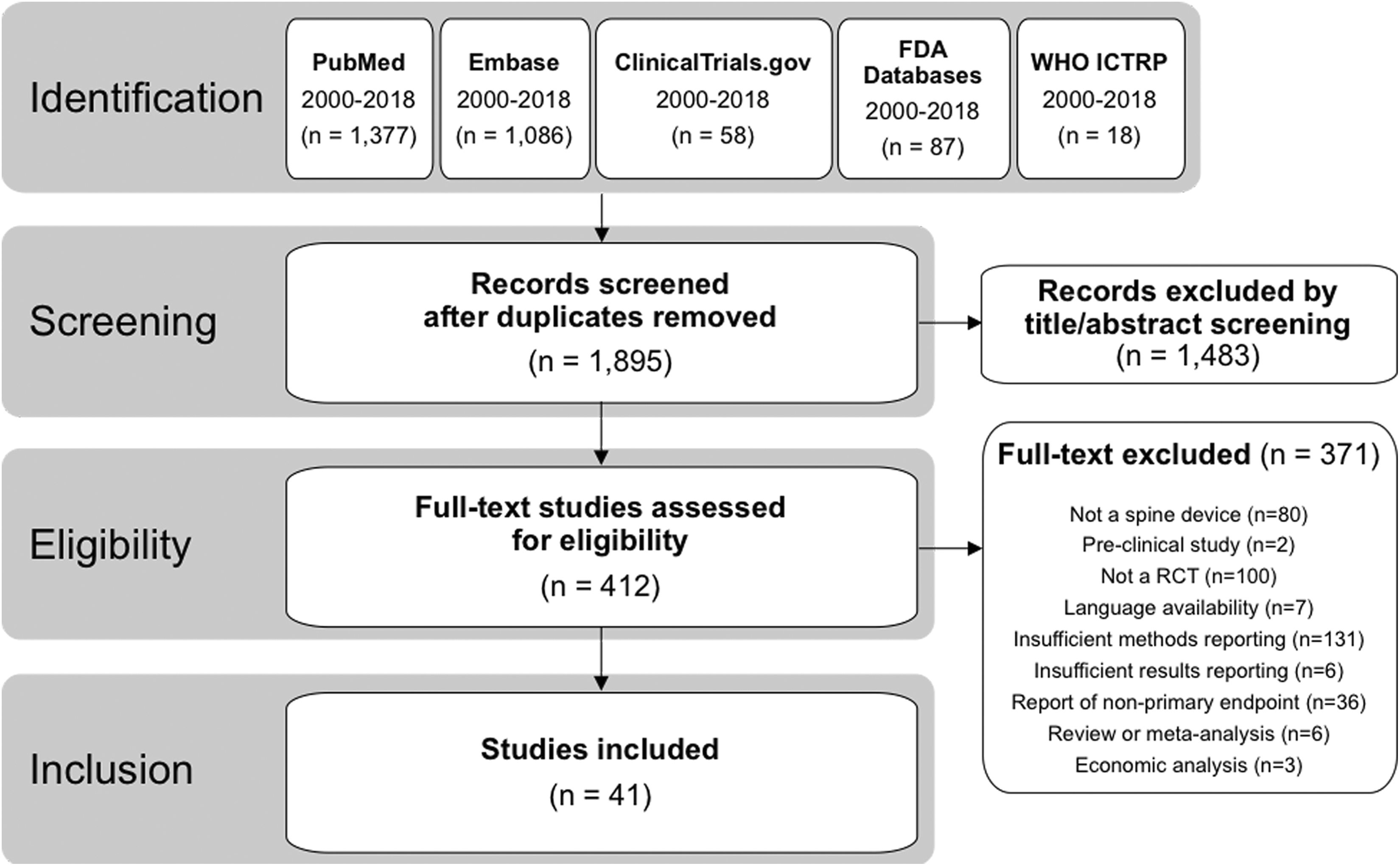
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) diagram demonstrating flow of records from identification through inclusion.
Summary of Included Studies.
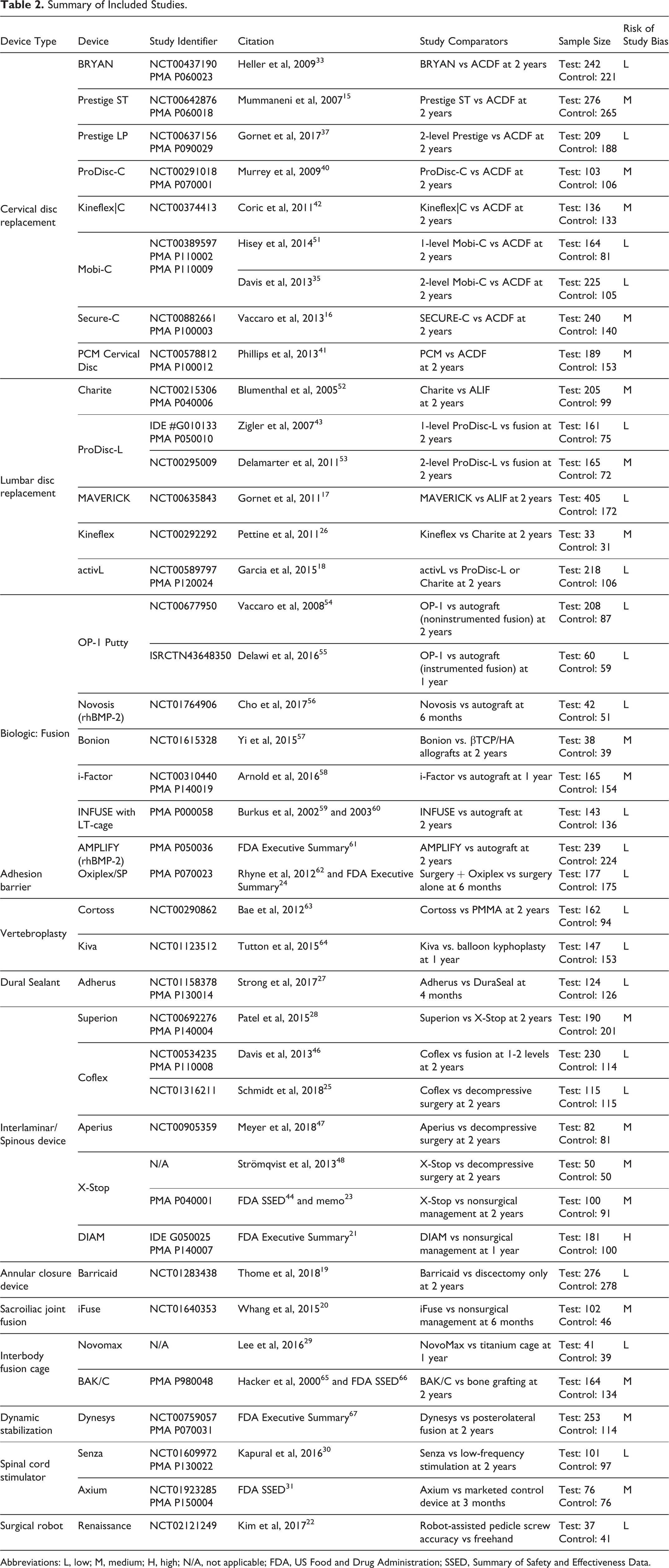
Abbreviations: L, low; M, medium; H, high; N/A, not applicable; FDA, US Food and Drug Administration; SSED, Summary of Safety and Effectiveness Data.
There were 19 (46%) studies designed as NI trials, 15 (37%) studies designed as NI + S trials, and 7 (17%) studies designed as superiority trials. Five of the 7 superiority trials were reported within the past 3 years (Figure 2). A composite clinical success (CCS) criterion was the most common primary outcome measure and was typically defined as: an improvement in a patient reported outcome greater than a clinically relevant threshold; the absence of secondary surgical interventions or procedures; the absence of neurologic deterioration; the absence of device and/or procedure related serious adverse events; and possibly radiographic findings. 12
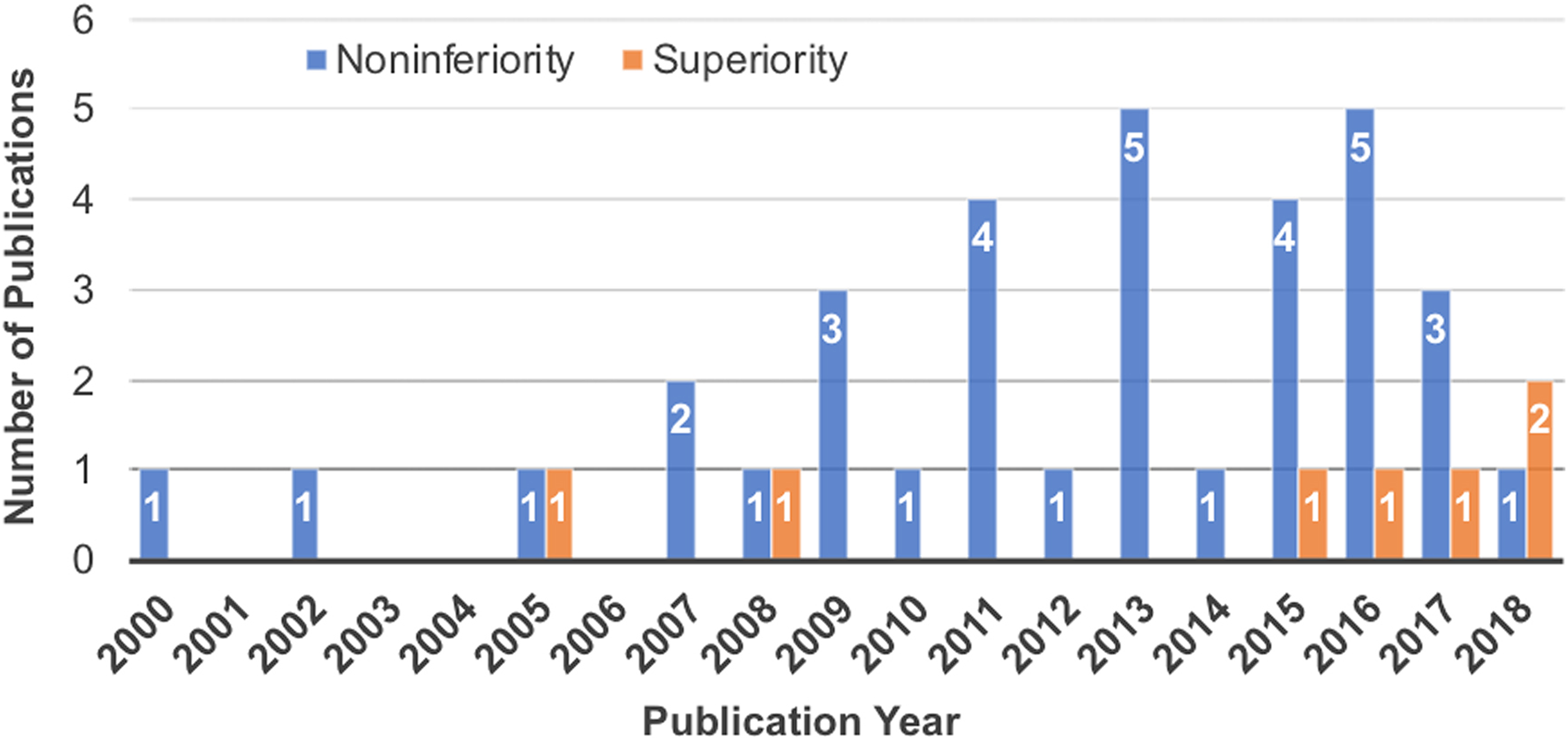
Trends in noninferiority versus superiority designs for randomized controlled trials of spinal devices that were included in this review since the year 2000.
Sample size calculations were often performed using the methods described by Blackwelder et al 13,14 with a NI margin of 10% (Table 3). A few studies assumed other margins for power calculations, but data was also analyzed with a 10% margin at the request of the FDA. 15 -18 No study estimated the NI margin from a prior superiority study that measured the effect size compared with sham or placebo. Three of the superiority studies used Bayesian methods for sample size. 19 -21 Two assumed superiority effect sizes of 9% 22 and 23%. 23 One did not describe its power analysis 24 and one assumed a medium effect size (Cohen’s d=0.4) for differences in disability scores. 25 Three superiority studies compared with nonsurgical management, 20,21,23 while the rest of the studies used an active surgical control. The active surgical controls represented a standard treatment technique for the respective condition (eg, fusion as a control for disc replacements and autograft for biologics). Seven of the studies compared with devices of the same class that were already available on the market. 18,26 -31
Summary of Study Design Characteristics and Conclusions.
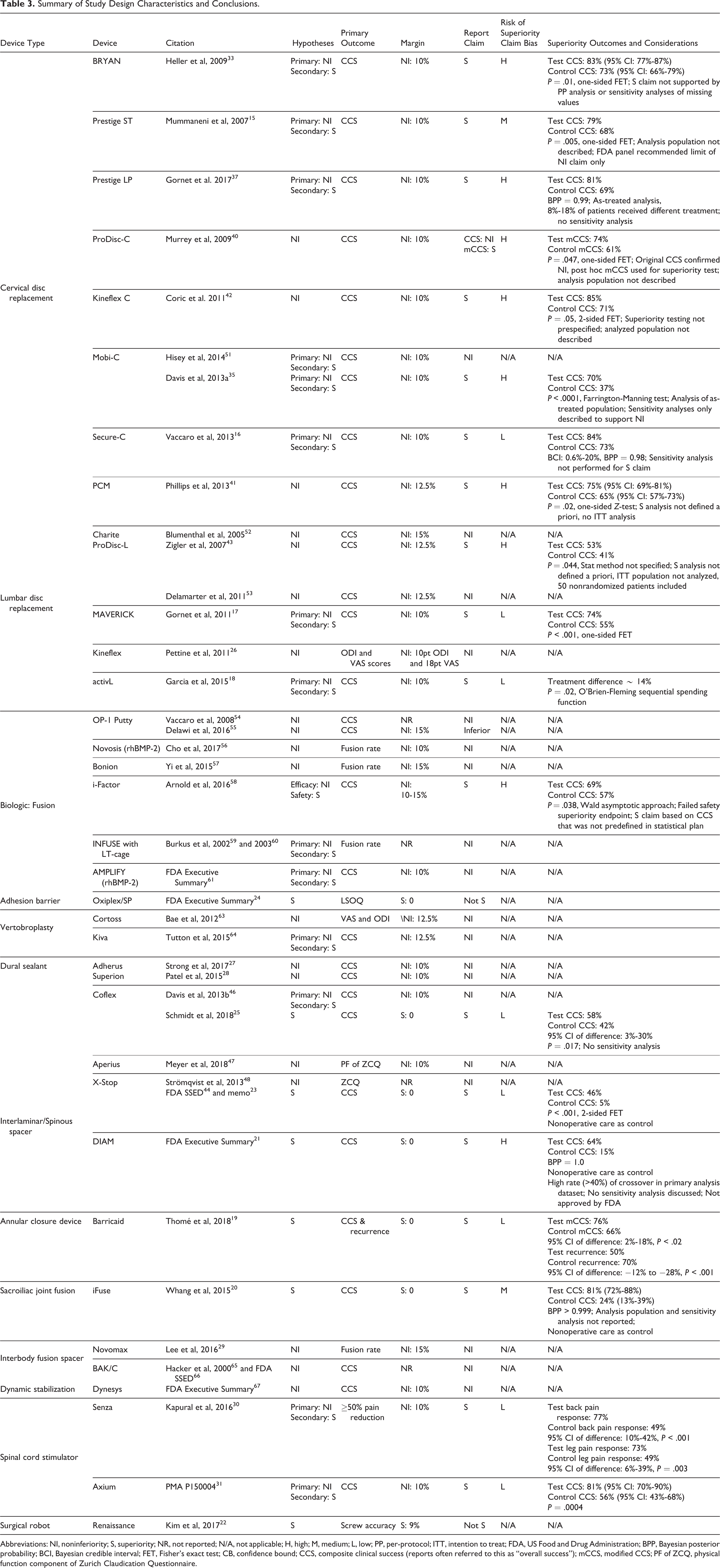
Abbreviations: NI, noninferiority; S, superiority; NR, not reported; N/A, not applicable; H, high; M, medium; L, low; PP, per-protocol; ITT, intention to treat; FDA, US Food and Drug Administration; BPP, Bayesian posterior probability; BCI, Bayesian credible interval; FET, Fisher’s exact test; CB, confidence bound; CCS, composite clinical success (reports often referred to this as “overall success”); mCCS, modified CCS; PF of ZCQ, physical function component of Zurich Claudication Questionnaire.
The overall risk of study bias was low in 22 of the 41 studies (54%), medium in 18 (44%), and high in 1 (2%) of the studies (Table 2; Supplementary Table S1). Medium risk ratings were attributable to potential attrition bias, an unclear blinding of outcome assessors, or potential limitations in randomization. The study with a high risk of bias suffered from a high rate of crossover subjects in the primary analysis dataset, a lack of sensitivity analyses, and uncertainty of concurrent interventions that could confound outcomes. The use of independent assessors, such as radiologists who were blinded to other outcomes, was considered an appropriate substitute for investigator blinding. Financial disclosure statements were only provided in 23 (56%) of the reports.
Evaluation of Superiority Claims
Among the 19 NI studies, 4 (21%) made post hoc superiority claims. Ten of the 15 (67%) NI + S studies and 5 of the 7 (71%) superiority trials satisfied their a priori superiority hypothesis (Table 3). All 19 superiority conclusions were based on statistical analyses with a superiority margin equal to zero. Although none of the studies discussed the superiority margin, the difference in proportions of treatment success (CCS) exceeded a +10% margin in 16 of the 19 studies (Table 3). However, the lower bound of the 95% confidence interval did not exceed +10% in most of the studies reporting that information.
The superiority claims in 4 of the 10 NI + S studies were found to be at a high risk of bias and 1 was at a medium risk of bias (Table 3; Supplementary Table S2). The NI + S study with a medium risk of bias for the superiority claim did not describe the analysis population and the FDA panel recommended only allowing NI claims. 15,32 In 1 NI + S study at a high risk of bias, the superiority claim was not robust to the sensitivity analyses of imputed values or the per-protocol analysis. 33,34 Two other NI+S studies at high risk of bias performed the superiority analysis on the as-treated population rather than the ITT population and did not describe any sensitivity analyses for the population or missing value imputations. 35 -38 This is particularly important when up to 18% of patients did not receive the assigned treatment, which could compromise the efficacy of randomization. 37 The fourth NI + S study at high risk of bias only reported the safety analysis to be a predefined superiority analysis, which failed to meet statistical superiority; yet, overall success rates were claimed to be superior. 39
All 4 superiority claims from NI studies were rated to be at a high risk of bias due to the apparent post hoc specification of the superiority hypothesis and lack of multiplicity adjustment. 40 -43 Furthermore, the analysis population was either not described 40,42 or the per-protocol population was used 41,43 in each of these studies. One NI study claimed superiority based solely on a post hoc modified CCS outcome since only NI could be claimed with the original primary endpoint. 40
Three of the 5 superiority trials were rated as a low risk of bias for the superiority claim, 19,23,25,44 1 was rated with a medium risk of bias due to the lack of reporting on the analysis population or sensitivity analyses, 20 and 1 was at a high risk of bias due to a high rate of crossover in the primary analysis dataset, no sensitivity analyses, and potentially confounding concurrent interventions. 21 The study at high risk of bias did not lead to FDA approval of the investigational device. Among these 5 superiority trials, only 2 described sensitivity analyses (the conclusions were robust to the alternate analyses). 19,23 Although it was not considered in the risk of bias evaluation, 12 of the 19 (63%) studies claiming superiority reported the associated confidence intervals, which are useful for understanding the effect size. These 12 studies were comprised of 1 of the 4 NI studies, 7 of the 10 NI + S studies, and 4 of the 5 superiority studies.
Discussion
The majority of RCTs for spinal device trials are designed primarily as NI trials based on effect size, secondary benefits, or ethical considerations; however, sponsors frequently attempt to establish post hoc superiority claims. The present study demonstrates that post hoc superiority claims derived from NI trials often suffer from a high to medium risk of bias due to analyzing the per-protocol or as-treated populations without sensitivity analysis, the claims not being robust during sensitivity analysis, or the claim being based on post hoc modified endpoints. This is important, since sponsors are under pressure from physicians, payers, and health care systems to demonstrate improvements in safety, efficacy, and cost-effectiveness. By claiming superiority in some aspects of safety and effectiveness, the sponsor can argue an improved value proposition. The current study suggests that such post hoc claims may be valid in some instances but should be scrutinized closely by the intended audiences.
The strengths of the present study include the use of multiple databases, the inclusion of important governmental databases in addition to indices of journal articles, a rigorous query methodology, and the use of multiple reviewers to eliminate false positives and combine duplicates from the query results. However, there are several shortcomings to the results. No set of databases or queries can assure complete capture of relevant results. Also, many RCTs have multiple published reports at multiple follow-up timepoints. We focused on the timepoint for the trial’s primary endpoint; however, it is possible that additional superiority claims were made at later timepoints. Published protocols that provided adequate details of the a priori study plans were usually unavailable. A published protocol was only identified for 1 study. 19,45 Another limitation was that important details were sometimes not reported, which resulted in an “Unclear” rating for the bias assessments. Similarly, the disclosure of potential conflicts-of-interest was not consistent and could not be meaningfully collected and analyzed. While regulatory bodies and payers may receive additional, nonpublic details of the trials from the sponsor, other researchers must rely on publicly available data. Finally, only superiority claims related to primary outcomes at the primary endpoint were evaluated in this review; however, analyses of secondary outcomes specified a priori can be important for determining the utility of a new device, particularly for NI trials.
Only 17% of the reviewed RCTs of spinal devices since 2000 were designed as superiority trials. Major categories of NI trials included disc replacements (15 studies), biologics for fusion (7 studies), and interspinous/interlaminar spacers (7 studies). Most disc replacement studies compared with fusion, offering the secondary advantage of retaining range of motion. While some of these studies included radiographic measures of motion or fusion, they still used a NI design for the primary endpoint. Biologics studies had the secondary advantage of avoiding donor site morbidity compared with autologous iliac bone grafts, but this was not articulated as a superiority hypothesis and was only indirectly captured in patient reported outcomes described in the NI hypothesis. Interlaminar and interspinous process spacers are promoted as less invasive surgery, but only 1 trial compared an interlaminar device directly to fusion in order to justify the implication that reduced operating room time and blood loss resulted in a net benefit. 46 Other reports comparing interspinous process spacers to decompressive surgery alone referred to improving patient satisfaction, complication rates, and reducing subsequent surgical interventions for the potential advantages of the new devices. 47,48 Such comparisons would be most appropriate as a superiority trial with adverse events included in the CCS, as exemplified by Schmidt et al 25 for an interspinous process spacer versus decompression and analogously by Thomé et al 19 for an annular closure device compared with discectomy alone. Updates to the Consolidated Standards of Reporting Trials (CONSORT) statement were proposed in 2006 49 and incorporated in 2012, 50 which suggest that studies should report the rationale for adopting a NI design and the associated NI margin. Most NI or NI + S studies published after these updates did not specifically discuss rationale for NI vs. superiority designs. Furthermore, only 4 reports provided any rationale for the NI margin, referring to requirements by the FDA.
Using well-rounded CCS measures as the primary endpoint may reduce the options for secondary benefits of the device beyond possible economic advantages. Among the reviewed RCTs on disc replacement, the primary endpoint CCS rates in the control group (fusion) ranged from 37% to 73% 15,16,33,35,37,40 -42,51 for cervical discs and from 41% to 55% 17,43,52,53 for lumbar discs, suggesting that a ceiling effect should not be a concern in those studies. Yet each disc replacement was evaluated with NI as the primary hypothesis and superiority as the secondary hypothesis. By focusing on appropriate endpoints, at-risk populations, and CCS criteria that demand well-rounded device success, the ceiling effect can be diminished and areas for improvement can be elucidated.
This review observed four superiority claims made through post hoc analyses of NI trials. These superiority claims were inherently at a high risk of bias due to post hoc hypothesis specification in a confirmatory trial. 4 Furthermore, 50% of the superiority claims from NI+S studies were observed to be at a medium or high risk of bias due to inappropriate methodology for analysis or interpretation of the superiority hypothesis. This was usually attributable to analyzing the as-treated or per-protocol population without consideration of the ITT dataset. Relying solely on as-treated or per-protocol analyses could bias the conclusions, particularly if a significant number of patients did not receive the assigned treatment, there was missing follow-up data, or significant attrition. 7 Overall, such deficiencies were apparent in 64% (9/14) of the NI or NI + S studies making superiority claims, which demonstrates the challenge of ensuring high fidelity conclusions when the superiority hypothesis is secondary to the NI design.
Based on the studies reviewed herein alongside the theoretical considerations of trial design and interpretation, superiority claims derived from NI trials may have a greater likelihood of confounding by methodological mistakes, ambiguities or sources of bias compared to claims derived from superiority trials. However, RCTs with an NI + S design can indeed be rigorous and present superiority claims with high levels of confidence. A few of the reviewed NI+S studies had a low risk of bias in the superiority conclusion because of the meticulous nature of the analysis and reporting, which included sensitivity analyses of both the population dataset and missing value imputations along with confidence intervals that demonstrated substantial margins. 17,18,30,31 The rationale for conducting these studies as NI + S trials rather than focusing on superiority was unclear. Regulatory or commercial considerations may provide a possible explanation.
Conclusions
Spine studies rarely employ superiority designs for confirmatory trials. NI studies can sometimes yield reliable superiority claims, but meticulous study conduct, analysis, reporting, and interpretation is paramount. Considering the singular goal of superiority trials and the standard methodology of such designs, greater confidence may be derived more readily from the resulting superiority claims. Investigators and sponsors are encouraged to consider superiority trial designs when evaluating novel technologies against a standard of care when feasible. Readers are encouraged to carefully evaluate the risk of bias behind each superiority claim by examining the methodology of the study and associated analyses.
Supplemental Material
Supplemental Material, Appendices - Superiority Claims for Spinal Devices: A Systematic Review of Randomized Controlled Trials
Supplemental Material, Appendices for Superiority Claims for Spinal Devices: A Systematic Review of Randomized Controlled Trials by S. Raymond Golish, Michael W. Groff, Ali Araghi and Jason A. Inzana in Global Spine Journal
Supplemental Material
Supplementary_Table_S1 - Superiority Claims for Spinal Devices: A Systematic Review of Randomized Controlled Trials
Supplementary_Table_S1 for Superiority Claims for Spinal Devices: A Systematic Review of Randomized Controlled Trials by S. Raymond Golish, Michael W. Groff, Ali Araghi and Jason A. Inzana in Global Spine Journal
Supplemental Material
Supplementary_Table_S2 - Superiority Claims for Spinal Devices: A Systematic Review of Randomized Controlled Trials
Supplementary_Table_S2 for Superiority Claims for Spinal Devices: A Systematic Review of Randomized Controlled Trials by S. Raymond Golish, Michael W. Groff, Ali Araghi and Jason A. Inzana in Global Spine Journal
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: SRG reports personal fees from Intrinsic Therapeutics, during the conduct of the study; personal fees from US FDA, other from AAOS, personal fees from Paradigm Spine, and personal fees from Wright Medical outside of the submitted work. MWG reports royalty payments from Depuy Spine and Biomet Spine outside of the submitted work. AA reports personal fees from Intrinsic Therapeutics outside of the submitted work. JAI is a salaried employee of Telos Partners, LLC, which received consulting fees from Intrinsic Therapeutics during the conduct of this study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Telos Partners, LLC received funding from Intrinsic Therapeutics in support of the systematic literature review.
Supplemental Material
The supplemental material is available in the online version of the article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.