
Abstract
The approach used by medical information services in answering unsolicited safety-related questions from health care professionals regarding prescription medicines varies widely across the pharmaceutical industry. A significant amount of information is available in the public domain, but this can be difficult to filter and determine what is most appropriate for a given situation. A team representing the medical information group MILE (Medical Information Leaders Europe) and European Federation of Pharmaceutical Industries and Associations Pharmacovigilance Expert Group have partnered to develop principles and considerations on how to answer unsolicited safety questions. Essentially two key principles are important in ensuring success: (1) Effective collaboration between medical information and patient safety teams is important for an optimal outcome providing accurate, useful, and timely information. This article discusses considerations for an effective, efficient collaboration between medical information and patient safety and suggests a way of working. (2) Collaborating teams will need to evaluate and select the most appropriate sources of information to answer the question. Sources of information that may or may not be in the public domain are discussed. Adoption of principles and considerations discussed in this article may be expected to improve current safety information–sharing practices that tend to be conservative and risk averse. In addition, this presents the opportunity to initiate discussions with regulatory authorities to realize the benefits that will come through greater transparency and communication to support safe and effective use of medicines.
Introduction and Scope
Without context words and actions have no meaning at all.
Medical information (MI) and patient safety (PS) professionals working in the pharmaceutical industry today are very likely to connect with the above statement when considering the communication of safety information. In communicating medical information (including safety information), it is standard practice across the industry to provide accurate, factual, balanced information in response to unsolicited requests from patients as well as health care professionals (HCPs) and associated individuals or organisations. Pharmaceutical companies cannot provide regular proactive updates to HCPs on changes in safety information or context other than via updates to the Prescribing Information. These updates are provided to regulators in health authorities in the form of aggregate reports such as periodic safety update reports (PSURs).
The MI function within pharmaceutical companies responds to unsolicited requests for information related to companies’ medicines. Responses can be based on the Summary of Product Characteristics (SmPC) or based on other sources of information, which may or may not be in the public domain. In either case, the information provided must be nonpromotional and adhere to the principles outlined above.
Patient safety, also known as drug safety or pharmacovigilance, is the pharmaceutical science relating to the prediction, collection, detection, assessment, monitoring, and prevention of adverse effects with medicines. Safety departments are responsible for ensuring that the safety information available for HCPs and patients is consistent with the company’s position on the safety of their medicines. This company position, otherwise known as core safety information within the core data sheet, must be regularly reviewed and updated as appropriate, based on a comprehensive evaluation of all available information pertaining to possible adverse effects of a medicine. The value of this information in supporting the safe and effective use of medicines is entirely dependent on effective communication to HCPs and patients.
The fundamental source of safety information is the product label, and historically HCPs have supplemented this information with published scientific literature. Pharmaceutical companies, as developers, manufacturers, and/or marketers of medicines, have a large volume of information and a variety of sources, including information not approved under a medicine’s license (“off-label” information), from which they can draw answers to unsolicited questions. However, the industry is also highly regulated in what, how, and to whom they can communicate about the medicines they produce. In particular, information that can be provided to patients is limited to the product label and publicly accessible resources. Medical information cannot provide treatment advice to patients or HCPs. This means that the primary interaction of the pharmaceutical companies is with HCPs.
Within the European Union (EU), there have been 3 directives covering the advertising of medicinal products for human use. 1 –3 In addition to conforming to the EU Directives, companies voluntarily conform to the pharmaceutical industry codes of conduct, including those of the European Federation of Pharmaceutical Industries and Associations (EFPIA) as well as national pharmaceutical industry association codes of practice, all of which fit into the general framework of the EU Directives. 4
The approach to answering requests for safety-related information varies widely across companies with respect to the detail communicated and source documents used. As in many fields, the entrance of the Internet has transformed the MI space. Although it has increased transparency and allowed HCPs easy, rapid access to information regarding clinical trials and recommendations of regulatory authorities, the amount and quality of the information available online often presents a challenge. It can be difficult to filter large volumes of information for which context, quality, source, and reliability may be questionable. When such (mis-)information is used to make prescribing decisions, there is a high risk of a negative impact for patients receiving the medicine. 5
Every company with a registered medicine marketed in an EU country is required by law to provide MI services to answer HCP questions about their medicines. 2
Approximately 4 years ago, a group of European MI professionals identified that it is not always clear for HCPs how they can access such information from different pharmaceutical companies. This group has become a professional organization, MILE (Medical Information Leaders Europe), with the aim of facilitating access to information about medicinal products for HCPs and patients. 6 Easier access to high-quality information will support the safe and effective use of prescription medicines.
Members of MILE have partnered with PS colleagues to explore how to better respond to HCP expectations to provide comprehensive, reliable, and accessible safety information. With support from the EFPIA Pharmacovigilance Expert Group (PVEG), the output from this collaboration is formulated here. The objective is to provide principles and considerations for content and delivery of medicinal product–related safety information in response to unsolicited requests from HCPs to pharmaceutical companies. Central to these considerations are the collaborative working practices between MI and PS departments. Engagement with other internal stakeholders including medical affairs, product quality, regulatory, and legal may also be required.
The principles and considerations provided in this document apply to communications with HCPs. That includes any member, student, or researcher of the medical, dental, optometry, opticians, pharmacy, or nursing profession or any other person, social workers, clinical psychologists, formulary committee members, and pharmacy and therapeutics (P&T) committee members who in the course of his or her professional activities provides medical services and may prescribe, order, dispense, recommend, purchase, supply, administer, lease, or use pharmaceutical products and/or medical technologies, and all members of their office staff.
It does not relate to communications directed to patients, their relatives, or members of the public. However, it is expected that as companies consider adopting these principles, discussions will expand to include recommendations for improved communication with patients.
This document is not intended as a requirement. It is at the discretion of individual companies to decide how to apply these principles and considerations in line with their risk management strategy. The intention is to challenge current thinking in adapting processes to better meet HCP needs and optimize treatment decisions.
Considerations for the Development of a Collaborative Process Between MI and PS
When providing timely responses to requests for product-related safety information, both MI and PS will often be closely involved. The first step is to develop an internal process for handling responses to safety questions and establish a strong cross-functional working relationship. This project should be led by senior members of MI and PS together. If groups do not interact regularly, then establishment of a small internal working group is highly recommended to drive better collaborations.
To ensure successful cross-functional collaboration, MI and PS need to understand the role, responsibilities, and structure of both departments. PS may not be aware of the ways of working and restrictions under which MI groups operate. Likewise, MI are often unaware of the sources of safety information available to PS and the legal consequences of communicating them externally. A partnership of this nature is a great opportunity for both departments to develop a much fuller understanding of each other’s expertise and how best to utilize this in the interest of meeting customer needs.
Points of Contact
It is essential to ensure that an accurate and up-to-date contact list for key MI and PS positions exists. This might include the following contacts: local market MI and PS, regional and global MI teams, and therapy area PS teams (safety physicians, PS scientists, etc). In the absence of a more formal process of escalation of safety questions, this list at least provides points of contact if inquiries are received. Within PS departments, such lists form the basis of a compliant pharmacovigilance system and should be readily available and kept up-to-date as required by health authorities.
Escalations
MI departments should have processes in place to escalate complex requests received by their “front-line” MI staff to other MI staff (often referred to as second-line MI) or other designated functions. This escalation process will vary depending on the organization of the company. In some cases, it may be a consultation between MI and PS or the medical lead. If such a process already exists, it can form the basis for handling safety-related questions. An example escalation pathway may look like that shown in Figure 1.

Escalation pathway example.
The effectiveness of this process relies upon appropriate triage and subsequent escalation decisions at each level of the organization. MI professionals are highly skilled in responding to inquiries and should be given autonomy to do so when appropriate. However, it will also be important that they receive direction or training on when they need to seek further guidance before responding. It will be useful to develop an algorithm that can cover the majority of safety question “types” that may be received, for example, when there is no information available for MI through the following sources: - product label, - existing standard responses, - information available through literature search, textbooks, or - guidelines from local authorities; when the requester specifically requests case reports, risk management plan (RMP), etc; when the inquiry is complex or sensitive and needs expert medical guidance; and when there is a patient involved with an adverse event that is life-threatening or serious.
Other Considerations
The process for the development of safety responses should include clearly defined roles, that is, response creators, reviewers, and who should communicate responses to HCPs, as well as the timelines for response. Providing responses to HCPs (especially related to safety information) is time sensitive. The urgency of the HCPs’ need for the information should be determined on a case-by-case basis, for example, is there an imminent treatment decision, which is relying on this information. Lengthy review cycles should be avoided where possible and both MI and PS need to prioritize these activities. A good practice is to implement regular monitoring of shared metrics and key performance indicators. MI and PS need to align on specific situations and resources to be used for each product.
Questions and considerations that may help define the process for safety responses include the following: Do both parties understand the processes and limitations of the other party? Is PS management aware of MI initiatives and the importance of the role of PS in creating MI safety responses? Do PS staff routinely get feedback on what is done with the data/response they have provided? Consider a forum for exchange of information. Which safety-related standard responses should be reviewed by PS? All responses or just a selection? Which safety-related standard responses should undergo legal review? Who will develop responses? PS? MI? Who will review and communicate to HCPs? What can MI and PS expect from one another in terms of timelines for response? Inform PS upfront about upcoming revisions of existing standard responses for better PS resource planning. Are appropriate disclaimers agreed/available?
Principles for Responsible Sharing of Safety Information
Standard Practice for Medical Information Responses
MI responds reactively to specific questions from HCPs. They are not permitted to proactively provide information. Information may be within the terms of marketing authorization of the medicine (product license) or relating to off-label use or unlicensed medicines. In all cases, the answers provided do not go beyond the question asked while being balanced, factual, and evidence based. Answers are referenced, sourced from the highest levels of evidence available, and accompanied by appropriate disclaimers. In the provision of this information, data privacy and copyright laws are also adhered to. 7,8
Principles for Sharing Safety Information
When considering safety questions, the principles outlined above apply. In addition, the first consideration in responding to any question is to understand the exact question being asked, including whether this is associated with a specific patient adverse event. Any safety response should clearly state the source of the data provided, in addition to its limitations. Information provided should be based on evaluation by pharmacovigilance experts, including safety physicians within the company, and based on global safety databases rather than local as well as other relevant sources.
One of the most important elements in communicating safety information effectively and appropriately is providing context. The context of safety data needs to be clearly explained to ensure the HCP correctly understands the information and how it might apply to their patients. When benefit risk decisions are under consideration, context is essential to facilitate optimal patient outcomes. 9 See for an example on how to provide context to information.
The information communicated in safety responses should be based on clinical data and should include safety data from clinical studies as well as “real-life” safety monitoring data. Describing where uncertainty exists or where evidence is lacking can provide valuable information when making treatment decisions.
PS and MI teams in consultation with legal should agree on specific wording and disclaimer statements regarding caveats for interpreting safety data. This may require consultation with other stakeholders in the organization such as regulatory colleagues. Examples of disclaimers associated with specific categories of safety requests and some of the information sources are presented in Table 1.
Categorization of Safety Requests.
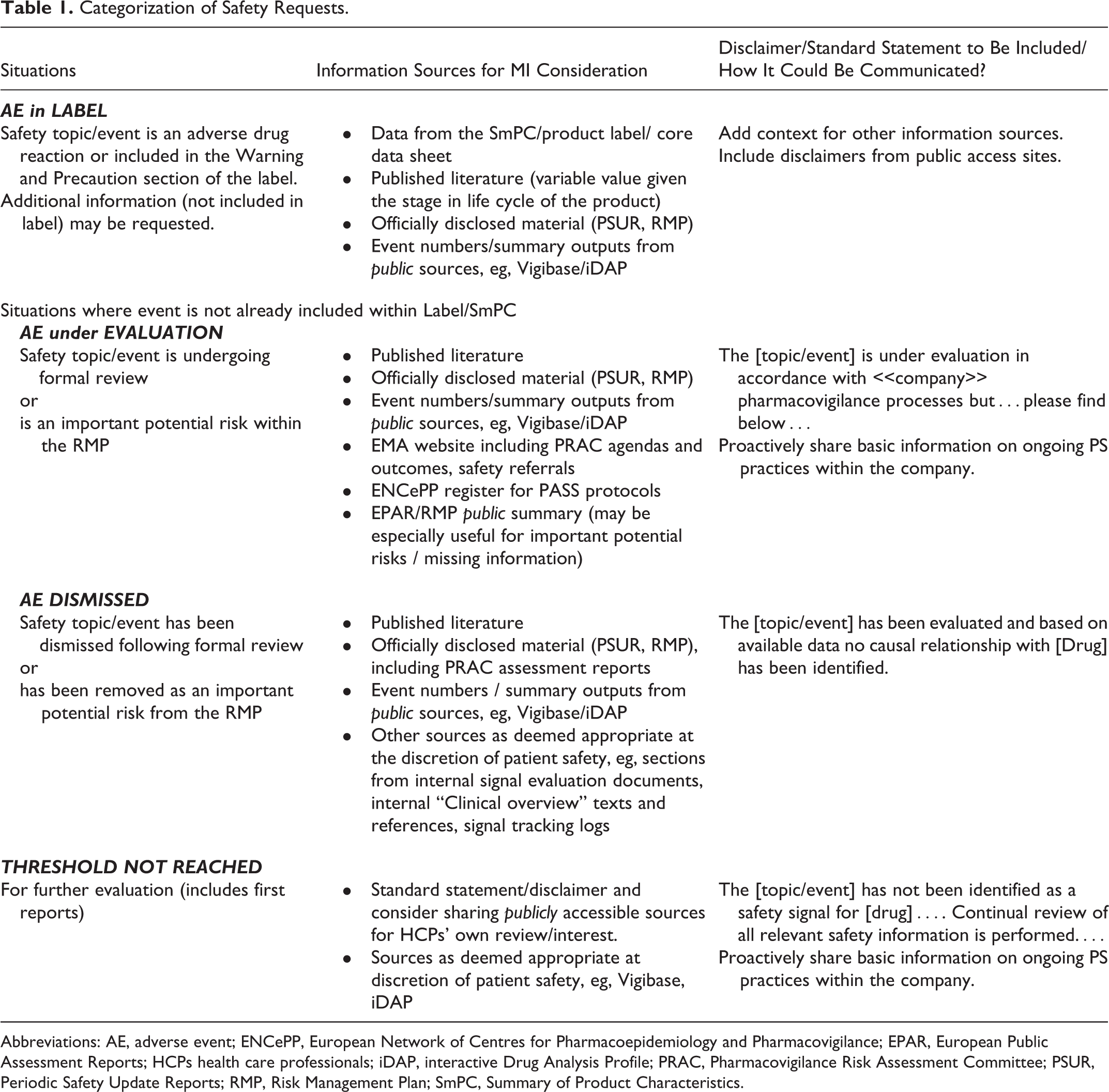
Abbreviations: AE, adverse event; ENCePP, European Network of Centres for Pharmacoepidemiology and Pharmacovigilance; EPAR, European Public Assessment Reports; HCPs health care professionals; iDAP, interactive Drug Analysis Profile; PRAC, Pharmacovigilance Risk Assessment Committee; PSUR, Periodic Safety Update Reports; RMP, Risk Management Plan; SmPC, Summary of Product Characteristics.
Example
Fictional Example to Illustrate Challenges and Opportunities with Safety Information Sources
A General Practitioner (GP) sends an enquiry to the MI department asking if Alopecia is a side effect of Treatment X.
A patient of his, a young lady in her mid-20s diagnosed with Multiple Sclerosis (MS) and started by Hospital Specialist on Treatment X, is anxious about possible Alopecia. The patient had quoted discussions on a patient forum that others have reported Alopecia and she has brought a screenshot from iDAP Figure 2.
The GP contacts the company MI department and asks for advice on how to address concerns of his patient. He mentions that he has checked British National Formulary (BNF) but did not find reference to Alopecia. He has also copied the screenshot from iDAP and admits he is not familiar with these information resources.
What Response Can Be Provided to Address His Enquiry?
It is assumed that alopecia is not in Core Data Sheet (CDS) or local label.
Response Option 1
A relatively short response stating that Alopecia is not considered to be causally associated with Treatment X and that Alopecia can occur secondary to number of causes: increased association with Autoimmune disorders for example. A publication on non-specific background rates is provided.
Response Option 2
As above but in addition: Detailed explanation of how the Company performs ‘routine pharmacovigilance activities’- (likely this could be provided based on existing wording from PSMF). Details of comparative frequency from pivotal trials of Alopecia (may well be available in published CSRs on EMA web site)
Both responses are possible and correct. When answering questions consideration should be given as to which option will help HCPs make informed decisions. In addition, these efforts will support building trust in pharmaceutical companies.
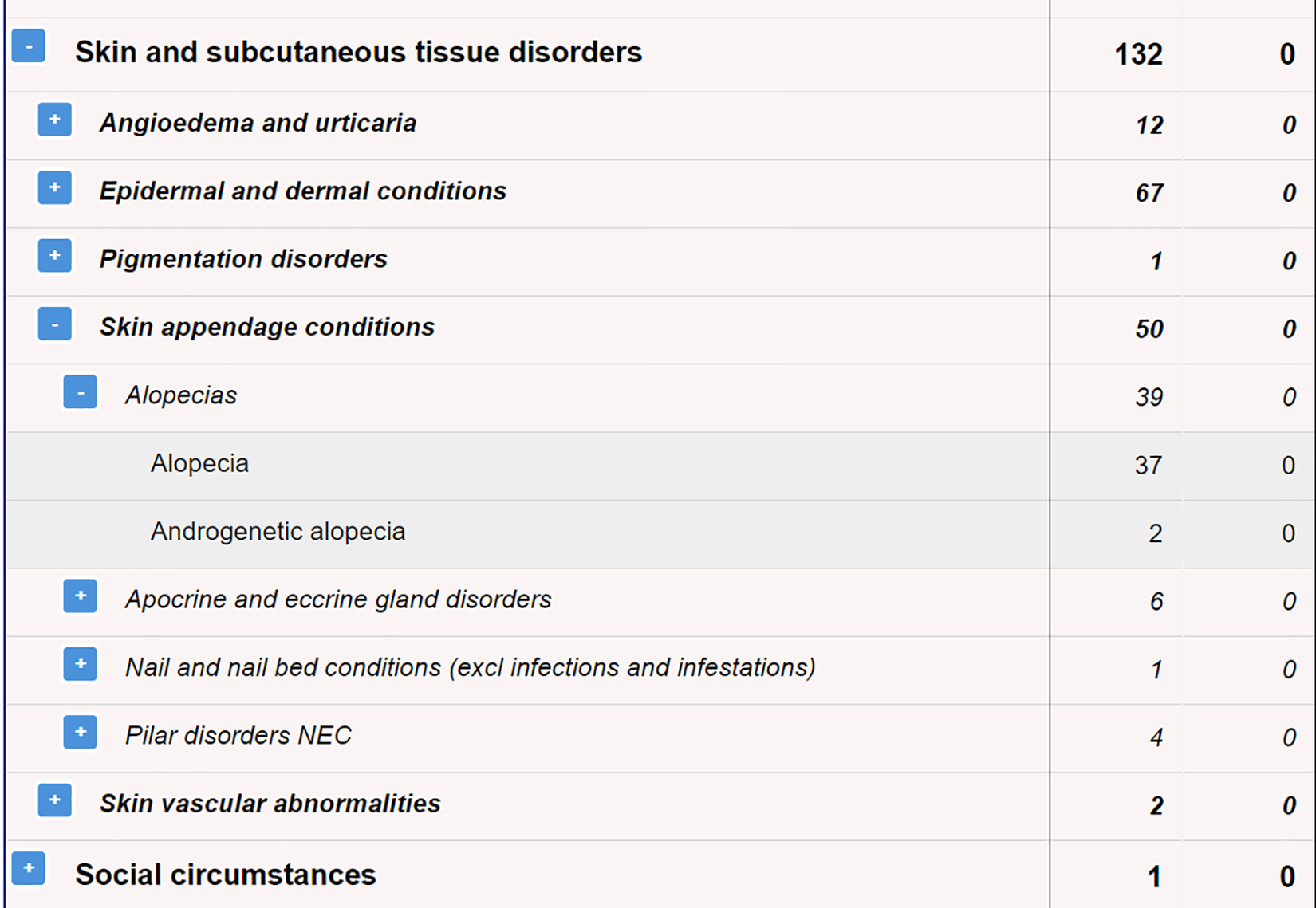
Screenshot from iDAPs provided by patient.
Selection of Appropriate Safety Data and Resource
The approach proposed here applies to situations when information is not directly available in the product label or published literature and when HCPs specifically request this information. In determining what information should be shared to answer safety-related questions, PS and MI colleagues need to gain agreement with senior medical, safety, and legal managers on what resources are appropriate to share. Discussions should consider what information is currently available in the public domain and what further information from internal data would be beneficial and possible to provide. The following questions and considerations may be helpful to support the discussions: What information is included in the SmPC / product label / core data sheet (considered the first resource of information)? Are there any other useful information sources that could help the HCP to use the product appropriately? What are the available public reference sources that MI and PS need to search and summarize or even share with HCPs? These are typically reviewed by PS as part of routine pharmacovigilance, but knowledge of their presence and potential value vary. Examples of resources in the public domain include:
-
-
-
-
-
-
-
-
-
-
When utilizing these sources to support responses, the limitations to the data they provide should be taken into consideration. For example, the EudraVigilance public access website presents the number of adverse events reported. These data do not include any assessment of a causal relationship to the suspected medicine and, therefore, should be viewed in this context. The majority of these public sources (eg, MHRA iDAPs, EudraVigilance) provide appropriate caveats and/or disclaimers to the viewer. It is important that these are relayed to the recipient of the information to help them put the information into the appropriate context.
What other nonpublic resources could be considered? - Much information is not fully accessible today but could be useful if provided with appropriate context. Data such as those from RMPs and aggregate reports (such as PSUR, development safety update report [DSUR], Periodic Benefit-Risk Evaluation report [PBRER]) offers significant additional insight to those with appropriate expertise. Sharing this requires careful consideration by pharmaceutical companies. - In-house data, for example, clinical data from study reports, signal evaluation reports, or position papers (data on file)
When sharing information from different sources as described above, the following should be considered: The recommended approach whenever possible is to extract data from the sources and summarize it in a response document providing context and including appropriate disclaimer statements. Avoid going beyond the questions asked. This may occur when sharing complete documents. Care should also be taken that information shared does not contain sensitive or confounding material. Direct the inquirer to the section in the document containing the information of interest when complete document is provided. When providing links to documents include additional context or explanation to help the reader understand the information, as well as disclaimer statements that discuss the caveats for interpreting safety data. Ensure compliance with data privacy laws where individual patient data is utilized. Most importantly, it is essential to always consider the risk for the patient or the company if the additional information is used inappropriately or out of context. Legal counsel can be sought in some situations according to company practices. Despite national codes of practice being aligned with EU directives and EFPIA code of practice some countries may have specific legal or regulatory requirements. Therefore, awareness of the country-specific requirements and consultation with stakeholders at country level is essential.
MI safety requests may be considered under 4 different categories. Table 1 describes these and provides examples of appropriate sources of information and accompanying statements. The Appendix provides a practical example of how suggested sources of information may be used. The exact resources and text of statements should be agreed within a cross-functional team including MI, PS, legal, and other appropriate functions.
Conclusions
This article discusses principles and considerations that pharmaceutical companies can follow to facilitate responsible sharing of safety-related information through reactive communications with HCPs. Adoption of these principles is expected to improve current safety information-sharing practices that tend to be conservative and risk averse. Existing practices have not evolved at the pace of technological advances or changes in customer expectations. Therefore, challenge and evolution of current practice is required and will lead to more transparent and contextualized sharing of safety information to reinforce pharmaceutical companies as a trusted source of information for HCPs. The main barrier to adopting these principles and considerations currently is the lack of specific guidance for medical information. In the absence of guidance companies are inclined to take a conservative approach that hinders the adoption of the principles and considerations presented here. This presents an opportunity to initiate discussions with regulatory authorities regarding current practices, and align with broader collaborative initiatives among industry, regulatory authorities, academia, patients, and other groups that aim to establish a common ground to improve transparency and communication. This in turn will benefit patients.
Supplemental Material
Supplemental Material, Appendix_Hristoskova_et_al_8May2019 - Principles and Considerations for Responsible Sharing of Safety Information Via the Medical Information Channel
Supplemental Material, Appendix_Hristoskova_et_al_8May2019 for Principles and Considerations for Responsible Sharing of Safety Information Via the Medical Information Channel by Sashka Hristoskova, James Milligan, Jan De Wit, Jukka Pesonen, Robyn Rennick and on behalf of MILE (Medical Information Leaders Europe) in Therapeutic Innovation & Regulatory Science
Footnotes
Author Note
The views discussed in this publication are the views of the MILE association and not necessarily the views of each individual member company of MILE.
Acknowledgements
The authors acknowledge the following colleagues for their valuable consultation and discussion during the preparation of this manuscript: Andrew Finlay, Astra Zeneca; Marianne Larsen, Astra Zeneca, and Jill Voss, Novartis Pharma AG.
Declaration of Conflicting Interests
SH is an employee and shareholder in Novartis and a shareholder in Alcon; JM is an employee and shareholder in Astra Zeneca; JdW and RR are employees and shareholders in GSK; JP is an employee and shareholder in Orion Corporation.
Funding
MILE covered the costs associated with publication of this article. No other financial support was received.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.