
Abstract
Background:
“Patient experience data” (PED) refers to the systematic collection of meaningful data relating to the experiences, perspectives, needs, and priorities of patients. PED can augment traditional clinical trial data in the FDA’s review of product applications. Section 3001 of the 2016 21st Century Cures Act requires the FDA to make a public statement about the PED considered in the approval of a drug application. Here, we present one of the first assessments of PED consideration during drug application approval, as reported by the FDA under Sec. 3001 of the Cures Act.
Methods:
FDA reported use of PED in the Review Documentation of the 59 new molecular entities (NMEs) approved in 2018 were collected, indexed, and cross-referenced against information regarding FDA review and product regulatory designation. The data reported in the PED tables were quantitatively described and visualized.
Results:
Of the 59 approved NMEs in 2018, 48 include a table that summarized whether PED was or was not used during the FDA drug review. Thirty-four of those 48 approvals (70.8%) reported using PED in the drug review. Patient-reported outcomes (PROs) represented the most significant source of PED and were used in 60.4% of approved drug reviews. Additional findings, including PED use by FDA review division and by FDA regulatory designation, are described.
Conclusions:
This assessment is a first step to better understanding how FDA considers PED in regulatory decision making. This analysis should help develop a baseline regarding FDA use of PED and may inform decisions to ensure patients’ experiences are adequately heard in future drug development.
Keywords
Introduction
Patient-focused drug development (PFDD) refers to a systematic approach to capture and meaningfully incorporate patients’ experiences, perspectives, needs, and priorities into drug development and evaluation. 1 PFDD recognizes that the patient is the ultimate expert in his or her condition and treatment, and thus can and should play an important role in understanding the burden of a disease and the benefit-risk profile of available treatments.
Patient experience data (PED) is a central component of PFDD and refers to the systematic collection of meaningful data relating to the experiences, perspectives, needs, and priorities of patients. This includes the collection of data concerning (1) the symptoms of their condition and its natural history; (2) the impact of the condition on their functioning and quality of life; (3) their experience with treatments; (4) input on which outcomes are important to them; (5) patient treatment preferences; and (6) the relative importance of any issue as defined by patients. 2 For example, PED can be included within a new drug application (NDA) or biological license application (BLA) to supplement clinical trial data with input from patients that would be collected and submitted by drug companies, caregivers, research teams, and disease-focused groups.
The US Food and Drug Administration (FDA) has undertaken multiple initiatives to bolster PFDD and use of PED in drug development and evaluation. FDA conducted 25 disease-specific PFDD meetings from 2012 through 2018 to more systematically obtain the patient perspective on specific diseases and their treatments. 3 Since 2015, FDA has supported and provided guidance to patient organizations to enable them in holding their own patient-focused externally led PFDD meetings concerning disease areas not yet examined. 4 FDA has also provided parameters for drug developers for qualification of tools for collection and regulatory application of patient-reported outcomes (PROs) through published guidance on PROs 5 and the Qualification Process for Drug Development Tools. 6
In 2016, FDA’s efforts were further supported by a provision in the 21st Century Cures Act (Cures Act) 7 and by FDA’s commitments made following the 2017 approval of the sixth reauthorization of the Prescription Drug User Fee Act (PDUFA VI). 8,9 In particular, Section 3001 of the Cures Act (Sec. 3001) requires FDA to publicly report PED considered in the approval of a drug application submitted after June 16, 2017, under the 505(b) and 351(a) pathways; the Cures Act does not specify the format or location in which the brief statement will be communicated to the public. 10
Here, we present one of the first assessments of FDA reporting of PED under the provisions of Sec. 3001 of the Cures Act. In this analysis, we first curate data relating to FDA use of PED during the 2018 review and approval of novel drugs under Food, Drug, and Cosmetic Act (FD&C Act) section 505(b) and Public Health Services Act (PHSA) section 351(a). We further reviewed these data to identify any possible trends across FDA review division and special regulatory designations. This analysis is a first step to better understanding the impact of PED on FDA regulatory decision making.
Study Data and Methodology
FDA has implemented the Cures Act Sec. 3001 requirement through the inclusion of a table (“PED table”) that indexes the types of PED considered. The PED table includes 18 separate check-boxes referencing nine forms of PED included by the sponsor in the submitted application, and three forms of PED not submitted by the sponsor but considered by FDA in drug review. Multiple boxes may be checked within a single table. When present, the PED table is included within the publicly available review documentation (“Review Documentation”).
Authors categorized the 18 check boxes of publicly available PED tables into three sections (Figure 1). Section A (boxes 1-12) notes PED that was submitted as part of the application and was considered by FDA in making an approval decision. Section A also includes a subsection of boxes (Section A1; boxes 3-6) that are dependent on box 2 and reference four forms of clinical outcome assessments (COA) used by FDA in application approval—Patient Reported Outcomes (PRO), Observer Reported Outcomes (ObsRO), Clinician Reported Outcomes (ClinRO), and Performance Outcomes (PerfO). Section B (boxes 13-17) indicates PED was not submitted by the sponsor but was used in making the approval decision. Section C (box 18 only) indicates that FDA did not consider any PED as part of the application approval. While the general structure, contents, and wording of the PED table is consistent between product approvals, there is some variation—for example, Sections B and C of the table are occasionally excluded if they are not applicable, and box 18 (indicating that no PED was used in application approval) may be replaced by a simple statement in the Review Documentation.
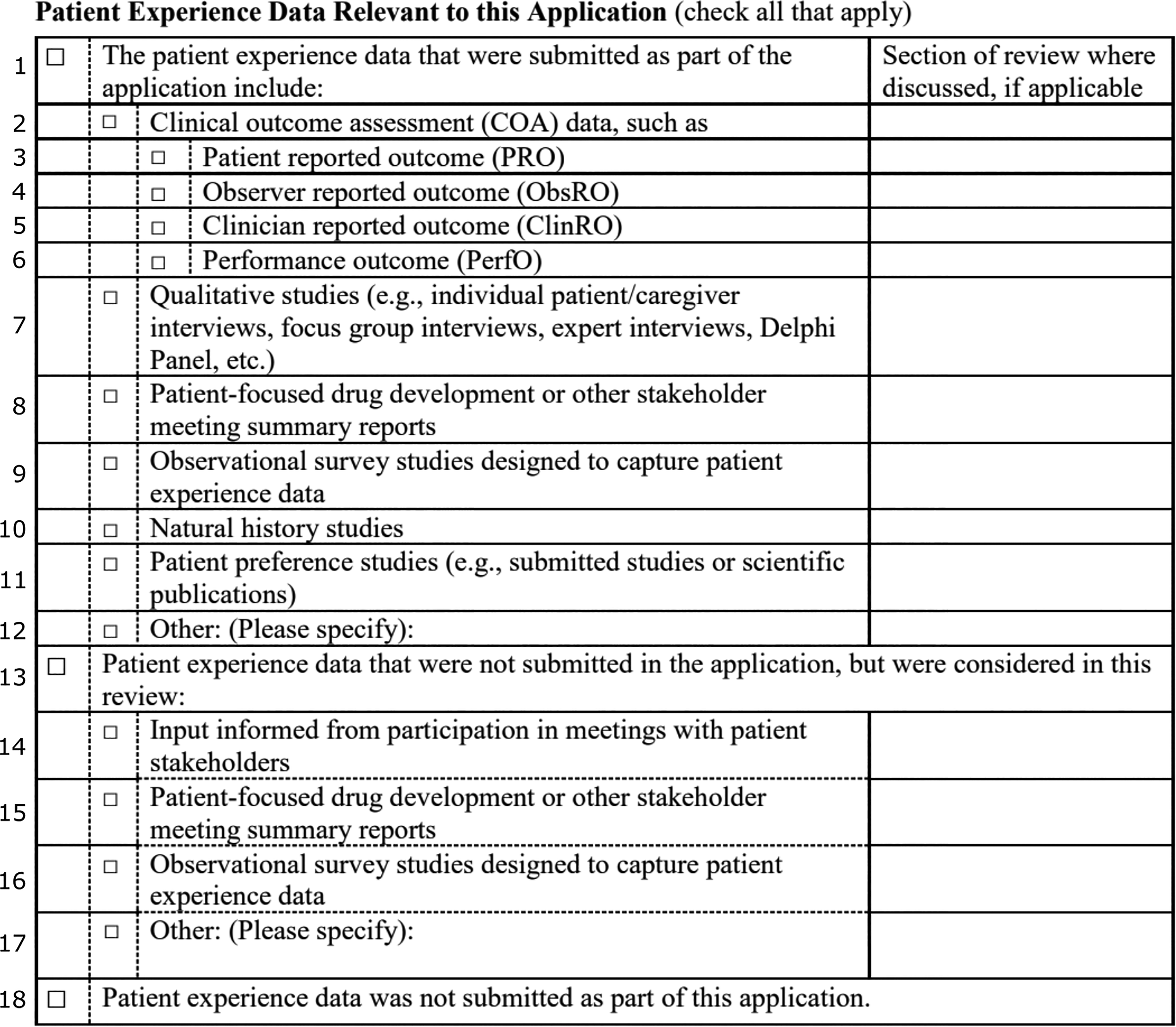
The Section 3001 PED Table with boxes numbered on the left. Section A includes boxes 1-12. Subsection A1 includes boxes 3-6. Section B includes boxes 13-17. Section C includes box 18 only.
Available PED tables for new molecular entities (NMEs) approved in 2018 were manually collected from the product Review Documentation. Although Sec. 3001 of the Cures Act may be interpreted to also apply to supplemental applications to an approved drug, PED disclosures for supplemental applications were not consistently publicly available and are therefore not included in the data set. Data collected for this analysis were corrected to account for box dependencies and inconsistency in FDA reviewer utilization of the table (eg, if box 3 is marked, then both box 2 and box 1 are considered to be marked as well; see Figure 1). The date of application submission and the review office and division were also collected from the Review Documentation. Information on special regulatory designations was extracted from CDER’s 2018 New Drug Therapy Approvals document. 11
The formatted data are presented in Supplementary Table S1. Analyses and data visualization were performed in R and Excel. 12 Curated data, data processing code, and analysis code are all available on GitHub. 13
Results
Forty-eight of the 59 drug applications approved by FDA/CDER (81.4%) in 2018 contained the Sec. 3001 PED table within the Review Documentation. Of the 11 that did not contain a PED table seven were submitted to FDA before the June 16, 2017, implementation date of Sec. 3001, and public reports of PED were not required. Two applications submitted prior to the implementation date of Sec. 3001 included the PED table. Four of the applications submitted after the June 16, 2017 implementation date did not include a summary PED table (DIACOMIT, KRINTAFEL, OXERVATE, and SYMDEKO). FDA reviewers of three drugs (TAKHZYRO, PALYNZIQ, XERAVA) had indicated that PED were used in the drug review (either box 2 or 13) but did not mark the specific type of PED used. The appropriate dependent boxes for these three drugs were adjudicated independently by two separate authors.
Thirty-four of the 48 approved drug reviews that included a PED table (70.8%) reported using PED in the review—29 drug reviews (60.4%) referenced sponsor-submitted data only, one drug review (2.1%) referenced nonsponsor-submitted data only, and four drug reviews (8.3%) considered PED submitted by both sponsors and nonsponsors.
Sponsor-Submitted PED
In total, 33 of the 48 approved drug reviews (68.8%) considered some measure of sponsor-submitted PED (Section A, boxes 1-9, Figure 2). Sponsor-submitted PED was primarily in the form of PROs—29 of 48 drug reviews (60.4%, box 3). Nine of these drug reviews indicated the specific PRO used. Other COAs were used much less frequently, and included ObsROs (2 of 48 approvals, 4.2%; box 4), ClinROs (8 of 48 approvals, 16.7%; box 5), and PerfOs (2 of 48 approvals, 4.2%; box 6). One product review (ONPATTRO) included COAs from all four categories.
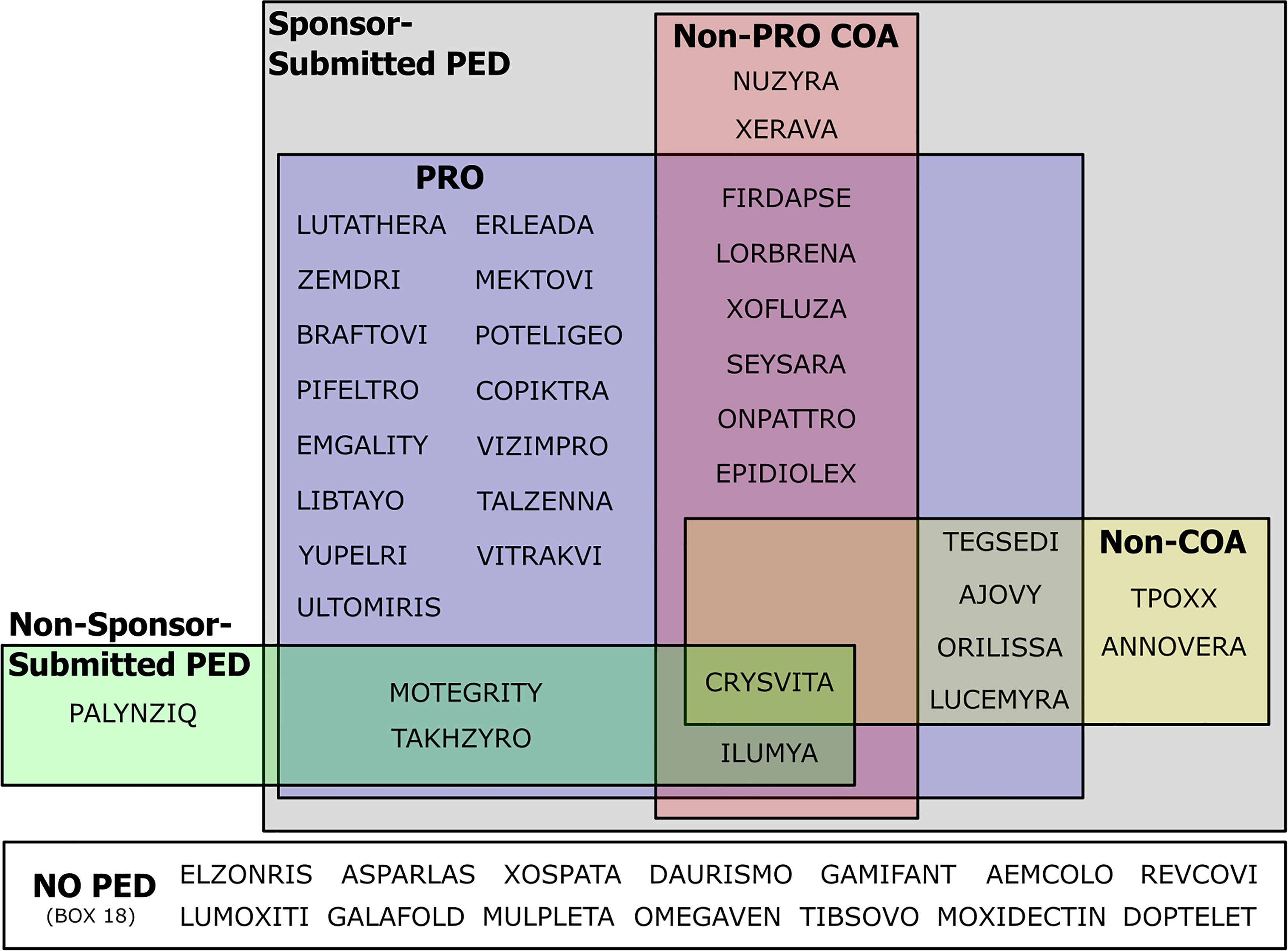
Diagram mapping the use of different types of PED in the Sec. 3001 tables for all novel approved products in 2018. COA, Clinical Outcome Assessment; PED, Patient Experience Data; PRO, Patient Reported Outcome.
Five approved drug reviews considered non-COA forms of sponsor-submitted PED—Qualitative Studies (ORILISSA, LUCEMYRA; box 7), PFDD Stakeholder meeting summary reports (TEGSEDI; box 8), Observational Survey Studies (CRYSVITA; box 9), Natural History Studies (CRYSVITA; box 10), and Patient Preference Studies (ANNOVERA; box 11).
PED Not Submitted by the Sponsor
FDA considered nonsponsor-submitted PED in five of the 48 approved drug reviews (10.4%). Four drug reviews (CRYSVITA, ILUMYA, TAKHZYRO, PALYNZIQ) considered information obtained from FDA PFDD meeting summaries (box 15), one product (PALYNZIQ) considered information from FDA participation in meetings with patient stakeholders (box 14), and one product (MOTEGRITY) indicated that PED from an Advisory Committee Meeting was considered during the review (box 17). Nonsponsor-submitted observational studies (box 16) were not used in any product approvals. Four of the five approved drug reviews that considered nonsponsor-submitted PED were reviewed by ODE III.
PED Considered Across FDA Review Divisions
We reviewed and categorized the use of PED across the various FDA Offices of Drug Evaluation (ODE) and review divisions (Figure 3).
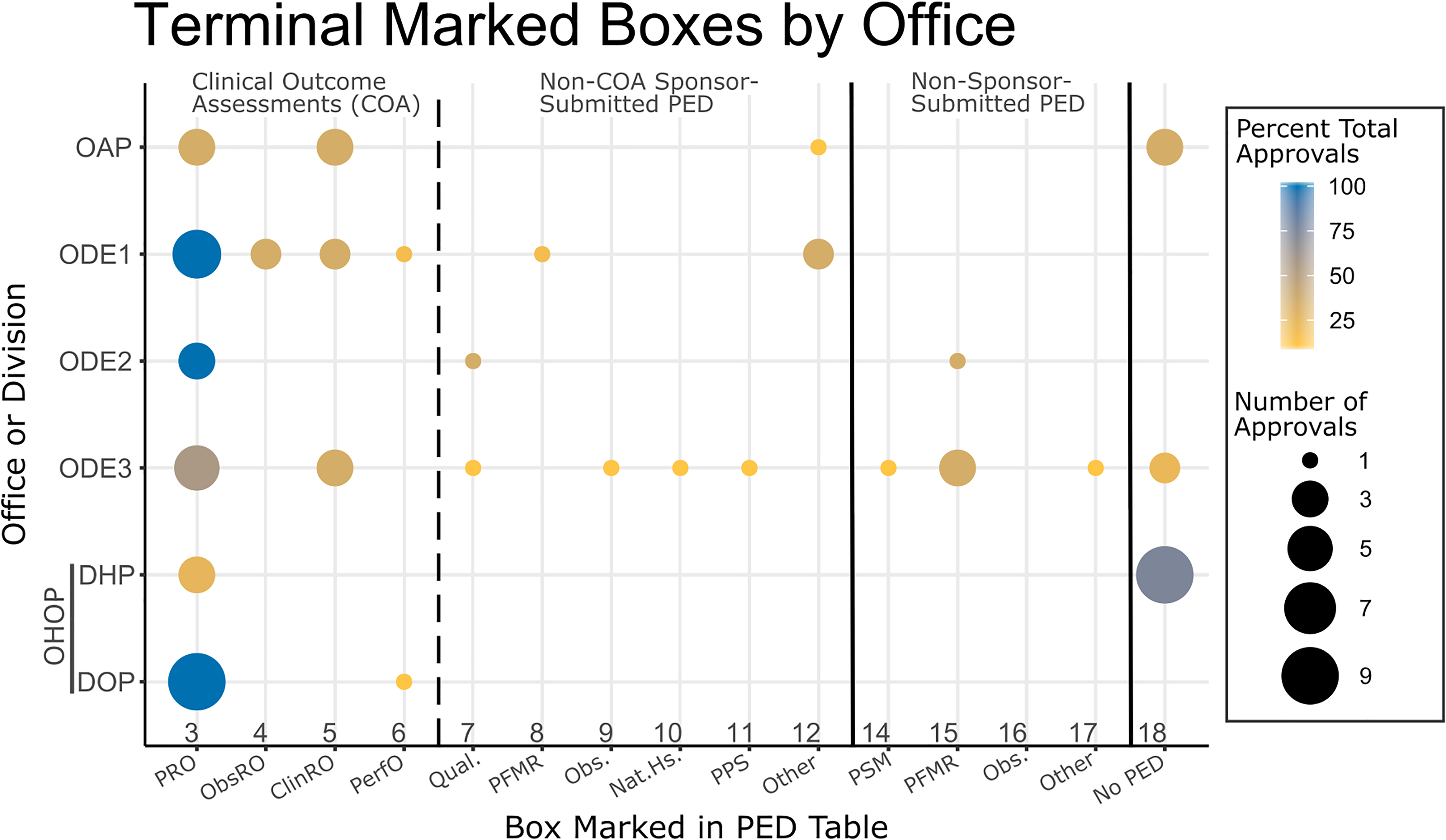
Breakdown of patient experience data (PED) inclusion by CDER Office. Only boxes that did not have dependent boxes (ie, “terminal”) are included. Dot area is proportional to instances of a box being marked in PED sections within a specific review office or division. Dot color indicates the percentage of total office or division reviews with that box marked. ClinRO, Clinician Reported Outcomes; DHP, Division of Hematology Products; DOP, Divisions of Oncology Products (both 1 & 2); Nat.Hs., Natural History Studies; OAP, Office of Antimicrobial products; Obs., Observational Surveys; ObsRO, Observer Reported Outcomes; ODE [1, 2, 3], Office of New Drugs; OHOP, Office of Hematology and Oncology Products; PerfO, Performance Outcomes; PFMR, Patient-Focused Meeting Reports; PPS, Patient Preference Studies; PRO, Patient Reported Outcome; PSM, Patient Stakeholder Meetings; Qual., Qualitative Studies. For full PED table box descriptions, see Figure 1.
PED Considered Across Special Regulatory Designations
Finally, we considered the use of PED in the review of products that received special regulatory designations from FDA. The small patient populations for orphan designated drugs and the shorter development and review times for other special regulatory designations could potentially increase the value of PED in these product reviews.
Many products received several special regulatory designations (Figure 4). Orphan drug designation (ODD) is assigned to drugs intended for the safe and effective treatment, diagnosis, or prevention of rare diseases/disorders that affect fewer than 200,000 people in the US, or that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug. 14 Breakthrough therapy designation (BTD) is assigned by the FDA to investigational drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s). 15,16 Fast Track Designation (FTD) is similar to BTD but does not require clinical data in order to receive the designation. 16 Accelerated Approval (AA) allows for approval of a drug product based on the drug’s effect on a surrogate endpoint, rather than a clinical outcome. 16 Priority Review (PR) shortens FDA’s review time from ten months to six months for products that demonstrate the potential to be a significant improvement in either safety or effectiveness. 16
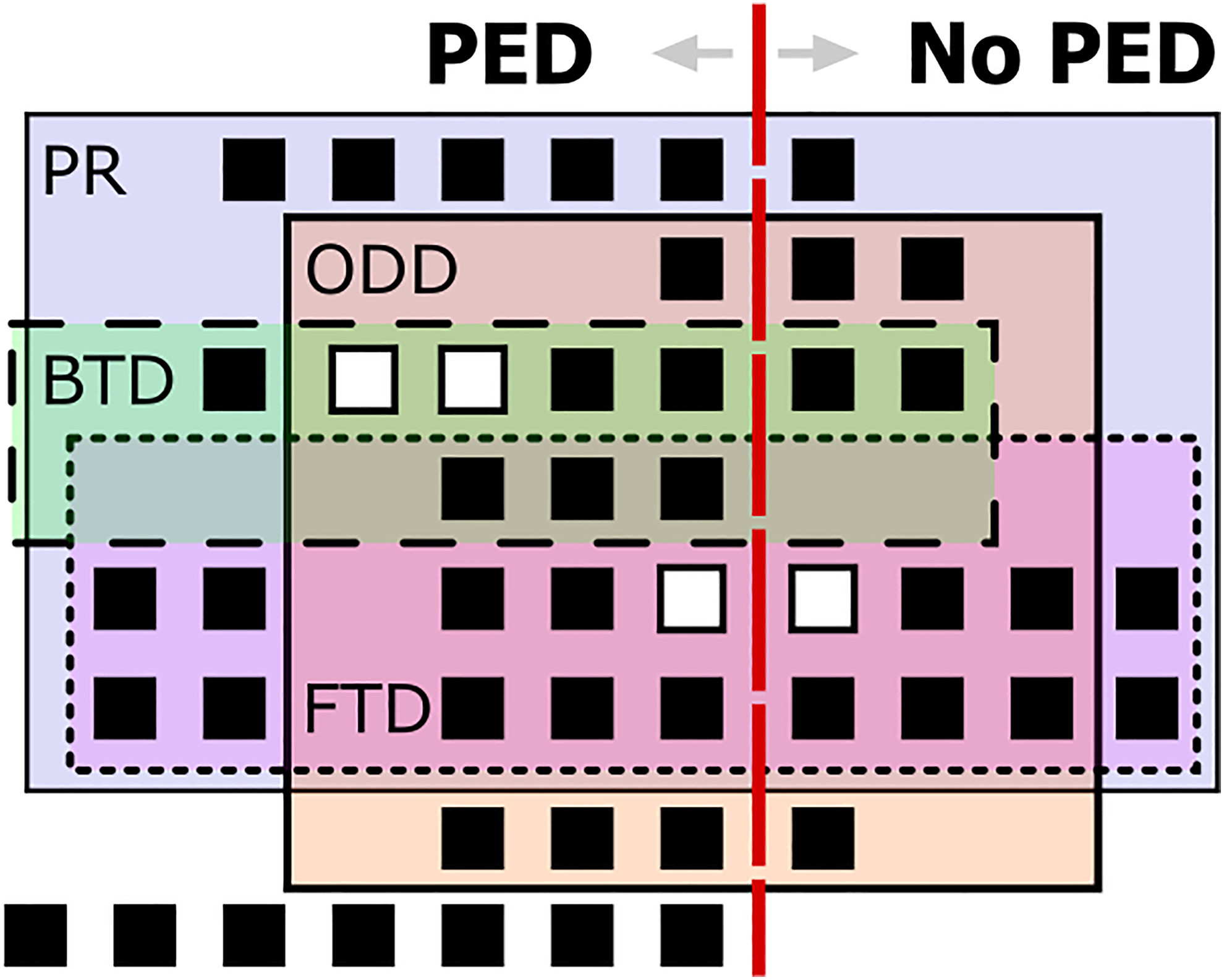
Diagram mapping the relationship of approved products that did or did not include patient experience data (PED) by special regulatory designation: AA, Accelerated Approval; BTD, Breakthrough Therapy Designation; FTD, Fast Track Designation; ODD, Orphan Drug Designation; PR, Priority Review. Each square represents a single approved product. Products that received Accelerated Approval are denoted as white squares.
Contrary to our expectations, product applications that received ODD were less likely to include PED within their Review Documentation. Seventeen of 28 (60.7%) approved orphan product reviews utilized PED, whereas 17 of the 20 (85.0%) approved non-ODD product reviews utilized PED (Table 1). Similarly, FDA considered PED for 13 of the 21 products that received FTD (61.9%); by contrast 21 of the 27 products that did not receive FTD utilized PED as part of their approval (77.8%). Twenty-four of 37 products that received PR utilized PED as part of their approval (64.9%); conversely, 10 out of 11 product reviews (90.9%) without PR included PED.
Percent PED Usage in Product Reviews With or Without Each Special Regulatory Designation.
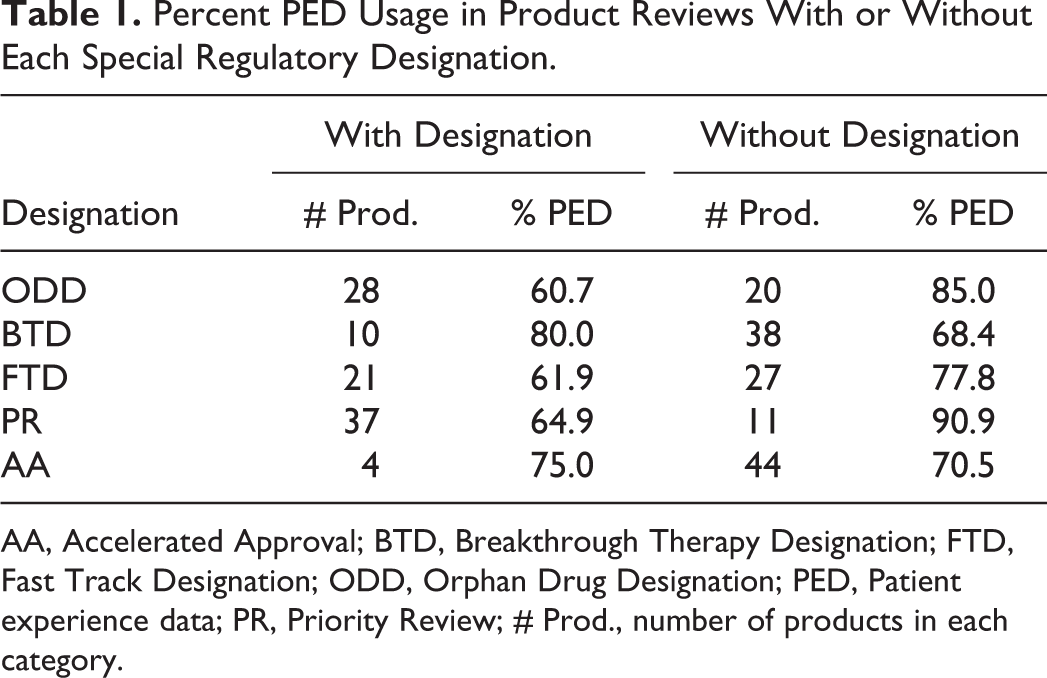
AA, Accelerated Approval; BTD, Breakthrough Therapy Designation; FTD, Fast Track Designation; ODD, Orphan Drug Designation; PED, Patient experience data; PR, Priority Review; # Prod., number of products in each category.
In contrast to ODD, FTD, and PR BTD and AA designated products were more likely to consider PED within their Review Documentation. Eight of the 10 products (80.0%) that received BTD also utilized PED during FDA review and approval; by contrast, only 26 out of 38 products (68.4%) that did not receive BTD included PED in their Review Documentation. Only four products were approved under the Accelerated Approval pathway, three of which utilized PED. Additional approvals of products receiving ODD, BTD, FTD, PR, and AA in the future may help to identify any statistically significant difference in PED use within these various regulatory contexts. Interestingly, all products that received a BTD either included PED within their product application or received an ODD (Figure 4).
Discussion
This report is an early analysis of the use of PED in FDA drug review and approval, as reported under implementation of Sec. 3001 of the 21st Century Cures Act. While FDA approved a record 59 novel drug applications in 2018, only 48 of those indicated whether PED was considered as part of the FDA application review. Although this sample analysis of drug approvals is relatively small, some general observations and trends suggested by the data are worth considering in the context of ongoing implementation of the statutory requirement.
First, FDA’s use of the PED table to fulfill the Sec. 3001 requirement is notable. Under the Cures Act, the HHS Secretary is required to “make public a brief statement regarding the patient experience data and related information, if any, submitted and reviewed as part of such application”; however, the placement (ie, where this public statement should appear) was not specified by law. Inclusion of the PED table within the FDA Review Documentation—which are by nature highly technical and accessed primarily by sponsors and academics—would limit their visibility to stakeholders less familiar with the FDA regulatory process (such as patient groups and healthcare providers).
Likewise, no specific form or format for the public statement required under Sec. 3001 was specified by law. Forty-six of the 50 product applications approved after Sec. 3001 implementation, and two products approved prior to Sec. 3001 implementation, included a PED table in the approval package. While the format of the table was largely consistent throughout these 48 drug product applications, the use of the table by FDA reviewers varied significantly—for example, FDA review teams in only 28 instances (58.3%) specified within the PED table the specific application section where the PED was discussed within the Review Documentation; in three instances, the table was incomplete, requiring independent assessment as part of our analysis; and the specific PRO instrument used was identified in nine of the 29 approved drug reviews that included PRO data. Further, it appears that FDA did not consistently publish a PED table for supplemental applications, which are also approved under FD&C Act 505(b) or PHSA 351(a)—while this information sometimes can be accessed by other means, there is no consolidated, “publicly available” access to this information.
In addition, FDA-approved drug reviews considered COAs (particularly PROs) more frequently compared to other more qualitative sources of PED. COAs accounted for almost three-quarters (73.2%) of the sources of PED included in approved novel drug reviews by the FDA, and PROs were the single greatest source (51.8%) of PED. This predominance of COAs and PROs could reflect several factors—for example, both the FDA’s and sponsors’ comfort and familiarity with use and application of PROs (as compared with other PED), given the more quantitative nature of PRO data and more historic use in a regulatory context. Regardless, other sources of PED, such as studies designed to gather patient input around disease or treatment burden, experiences during or after a clinical trial, patient preference, or other information gleaned from meetings with patient groups such as PFDD meetings and/or summary reports, appear to be considered less frequently by FDA in the context of drug application review. Based on the results of this review, both FDA and sponsors should ensure that all forms of PED, beyond COAs, are appropriately being incorporated into drug development for the purposes of FDA’s consideration in the context of review and approval.
Similarly, FDA might consider additional efforts to increase the diversity of PED sources. While FDA already encourages third-party patient groups to submit relevant PED and have committed to community outreach as part of the PDUFA VI Commitment Letter, PED was rarely considered during FDA review if not submitted by the sponsor (only 10.7% of PED sources were not part of the sponsor-submitted application). Further, the information from the 25 PFDD meetings held over the past 10 years was considered in only three drug application approvals (PALYNZIQ, TAKHZYRO, ILUMYA); three approved products—PIFELTRO (HIV 17 ), TALZENNA (Breast Cancer 18 ), and MOTEGRITY (Functional Gastrointestinal Disorders 19 )—were approved for indications addressed by FDA’s Voice of the Patient meetings, but did not report the usage of information from those meeting reports in their Review Documentation.
Finally, some possible correlations emerged that might warrant further exploration. For example, oncology product reviews almost exclusively used PROs; ODE III reviews were more likely to include nonsponsor-submitted PED compared to other CDER offices; products that receive ODD relied less heavily on PED within their applications; and products that receive BTD utilized PED at similar rates to non-BTD products within their product applications. If these correlations are real, they could be explained by several affecters, such as differences in study design between disease areas, the types of PED submitted by sponsors, culture of FDA review divisions, or the level of active engagement by the relevant patient associations or populations. The small sample size of products available for this analysis make definitive conclusions difficult; however, future tracking of these trends and their possible underlying causes may be worth monitoring.
Future Direction and Recommendations
We understand that FDA’s views on the statement of patient experience, and implementation of the requirements under Sec. 3001 continue to evolve, and that FDA staff is receptive to suggestions for improvement and advancement on the topic. In this analysis, the PED table was assumed to be an accurate reflection of the PED used during drug review. Given the incomplete nature of some of the PED tables, this assumption may not be accurate. Moving forward, we would expect FDA review divisions’ use of the existing table to become more standardized, both in terms of actual completion and in improving the linking of the PED referenced in the table to other sections of the Review Documentation (ie, rather than just noting a different section number, including a description of what aspects of review described in that section were affected by PED, and the result). Further, we contend that Sec. 3001 of the 21st Century Cures Act would include supplements to an approved application under 505(b) of the FD&C Act and 351(a) of the PHSA, and broader transparency into how PED is used in these contexts would be of great value to patients and drug sponsors.
Regarding the format of the public statement required under Sec. 3001, it would be useful to make information about FDA’s use of PED more detailed and accessible. One approach would be to include a patient-facing statement describing FDA’s use and application of PED for each approval—for example, in addition to the use of the Sec. 3001 table, publish a plain language accounting of its use of PED, explaining what PED were collected, how they were applied (ie, how FDA appraised this information and how that assessment affected overall benefit/risk review and approval, and potentially labeling), similar to the plain language descriptions of clinical trials used through the Drug Trial Snapshot initiative. 20 Likewise, making a public statement (via a PED table or other process) of the use of PED for supplemental product approvals would also be informative, and provide more examples of how FDA would consider and take into account this type of information. The public statement itself could be more detailed—for example, to the extent that COAs and PROs are included, an explanation of the tool and/or assessment (particularly when it is novel), or explaining how PED impacted the structured benefit-risk framework in the context of FDA’s review.
Finally, in order for all stakeholders to fully understand the import and impact of PED in the context of drug review, these more descriptive iterations (in both the Review Documentation and patient-facing materials) of FDA’s use of PED and/or COAs in drug review should be included in—or at least linked (eg, via QR code) to—the product’s approved labeling. Without direct inclusion in labeling, or at least a meaningful and accessible linkage of the information to labeling, true publication of FDA’s use of patient input arguably has not been effectuated for the benefit of all users. Standardization of the PED table, development of plain-text language documentation, and inclusion of PED in product labeling may more closely align the current PED reporting with the spirit and intent of the 21st Century Cures Act.
Supplemental Material
Supplementary_material_pdf - FDA Reported Use of Patient Experience Data in 2018 Drug Approvals
Supplemental Material, Supplementary_material_pdf for FDA Reported Use of Patient Experience Data in 2018 Drug Approvals by Cameron M. Kieffer, Alexis Reisin Miller, Benjamin Chacko and Andrew S. Robertson in Therapeutic Innovation & Regulatory Science
Supplemental Material
Supplementary_Table_1 - FDA Reported Use of Patient Experience Data in 2018 Drug Approvals
Supplemental Material, Supplementary_Table_1 for FDA Reported Use of Patient Experience Data in 2018 Drug Approvals by Cameron M. Kieffer, Alexis Reisin Miller, Benjamin Chacko and Andrew S. Robertson in Therapeutic Innovation & Regulatory Science
Footnotes
Authors' Note
This article does not contain any studies with human or animal subjects performed by any of the authors.
Acknowledgments
The authors would like to thank the many experts at Sanofi who provided comments on the manuscript.
Declaration of Conflicting Interests
Dr Kieffer is a PhRMA Foundation Regulatory Science Fellow working at Sanofi. Dr Robertson, Ms Reisin Miller, and Mr Chacko are full-time employees at Sanofi in the department of Regulatory Science and Policy, North America. Sanofi is a pharmaceutical company that develops and markets pharmaceutical products regulated by the FDA. The views expressed in the article are the views of the authors and do not necessarily reflect the views of Sanofi.
Funding
Dr. Kieffer's fellowship was supported by the PhRMA Foundation.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.