
Abstract
In the current pharmaceutical regulatory environment, patients continue to benefit from great advances in medical care. Sophisticated regulatory review systems have also evolved to ensure that safe and effective medicines are approved. However, these systems are not optimized in all countries. Gaps in individual regulatory agency capabilities together with duplication in non–value added national regulatory requirements, particularly in low- and middle-income countries (LMICs), can slow down regulatory approvals and therefore impede patient access to new medicines. These gaps exist despite the achievements in both regulatory convergence and harmonization of technical requirements by bodies such as the International Conference on Harmonization (ICH). There is a pressing need to strengthen regulatory review systems in emerging market economies as highlighted by the World Health Organization (WHO). These diverse challenges may seem overwhelming to individual national regulators, in part because of the sheer number of initiatives by multiple stakeholders, combined with a lack of information on concise practical actionable measures that can have a positive impact on review efficiency. This commentary presents 10 pillars that we believe represent the key hallmarks of strong regulatory review systems. Leveraging our internal company expertise at the global, regional, and country level across our entire product portfolio (both innovative and generic), we selected features proven to work in leading regulatory agencies, such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), which are also relevant for other regulatory authorities, especially in LMICs.
Introduction
Over the past several decades, advances in science and medicine have enabled patients to live longer and healthier lives than in the past. For example, HIV patients can now live relatively normal lives rather than facing a death sentence, rheumatoid arthritis patients have benefited from transformative biological and new small molecule treatments, and cancer patients are experiencing additional benefits as a result of advances in targeted therapies and immuno-oncology. 1 –3 Strong science-based regulatory review systems have provided a gateway for these medicines to reach patients, ensuring appropriate benefit-risk and increasing patients’ confidence that their medicines are safe and effective.
Significant advances in harmonization of technical requirements for development and registration of medicines have been achieved through platforms such as the International Conference on Harmonization (ICH) which supports efficient global development. 4 However, despite the expansion of ICH to include regulatory authorities in developing countries, regulators in low- and middle-income countries (LMICs) routinely face challenges in capacity, combined with gaps in capability, especially for new or specialized technologies. 5 From the perspective of a multinational company that operates in more than 125 countries and has hundreds of products in its portfolio, we have firsthand experience of some of the challenges that arise globally because of divergence in regulatory review requirements, particularly for non–science driven, non–value added factors. 6
The unintended consequence of the addition of nonessential requirements contributes to longer, less-efficient regulatory reviews that hamper the availability of medicinal products for patients even before considerations of affordability and access come into play. With this in mind, World Health Organization (WHO) resolution 67.20 is specifically directed at regulatory system strengthening for medicinal products and urges member states to undertake a series of measures to strengthen regulatory systems, for example “to identify the need to strengthen regulatory system capacity, collaboration and cooperation in the technically complex areas where substantial gaps may still exist.” 7
Many organizations are active in regulatory system strengthening activities including convergence and harmonization activities (eg, WHO, Asia Pacific Economic Cooperation [APEC], ICH, and Pan American Health Organization [PAHO]). 8 The sheer depth, breadth, and amount of activity in this area can be overwhelming. Against the backdrop of all these initiatives, we ask the question what practical steps can and should individual regulators undertake to ensure strong regulatory application review systems?
In this commentary, we propose 10 pillars that we believe represent the key hallmark features of strong regulatory review systems. The decision to focus on application review was deliberate in order to focus on actionable solutions for one aspect of regulatory strengthening. We decided from the outset to restrict the list to 10 features to drive identification and selection of items with the biggest impact, proven to work in leading agencies, for example, US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) and which are also relevant for other regulatory authorities, especially in LMICs (see Table 1). Inevitably, this is a generalized listing, not all features will be equally important to all regulatory authorities at a given point in time, and prioritization of these features will vary depending on the country. For these reasons, we have not presented these features in a prioritized order. Overall, we believe that the list as a whole represents essential features that characterize strong review systems (see Figure 1). The list of features was developed in an iterative manner within the company. The authors selected and proposed an initial list of key features based on their experience and expertise. The list was then further refined and finalized following consecutive reviews and feedback from senior regulatory colleagues experienced in working at the global, regional and country level and across business segments including both innovative and established medicines within the organization. Our proposed features are broadly consistent with a 2010-2011 Centre for Innovation in Regulatory Science (CIRS) survey on barriers and enablers in regulatory reviews. 9 Additionally, we believe they are complementary to the development of the WHO’s Global Benchmarking Tool (GBT). The GBT is a means by which WHO evaluates regulatory systems through a comprehensive and systematic benchmarking across 9 areas of regulation. Our specific focus on regulatory application review aspects may serve to highlight pragmatic ways to achieve strong regulatory review systems, thus supporting the broader initiative of the GBT. 10 Importantly, we see regular evidence that all these factors are valid via the reforms that the regulators themselves are undertaking. For example, in 2015 China initiated root and branch reforms seeking to put many of these features in place. 11 Even mature regulatory agencies need to constantly adapt to ensure that they can accommodate the latest medical and technological advances. For example, on June 4, 2018, the US FDA announced proposals to reorganize the Center for Drug Evaluation and Research Office of New Drugs (OND). Introducing a flatter structure with review divisions in additional therapeutic areas is expected to reduce management bottlenecks in performing application reviews while encouraging a stronger integration within and across offices via a concept termed as “integrated assessment.” 12 These examples support our belief in the value to all regulators from our proposed listing. Our hope is that by focusing on 10 features, this will provide a fresh impetus for aligned activity on strengthening regulatory review system requirements.
Summary of 10 Pillars and What These Look Like in Practice.

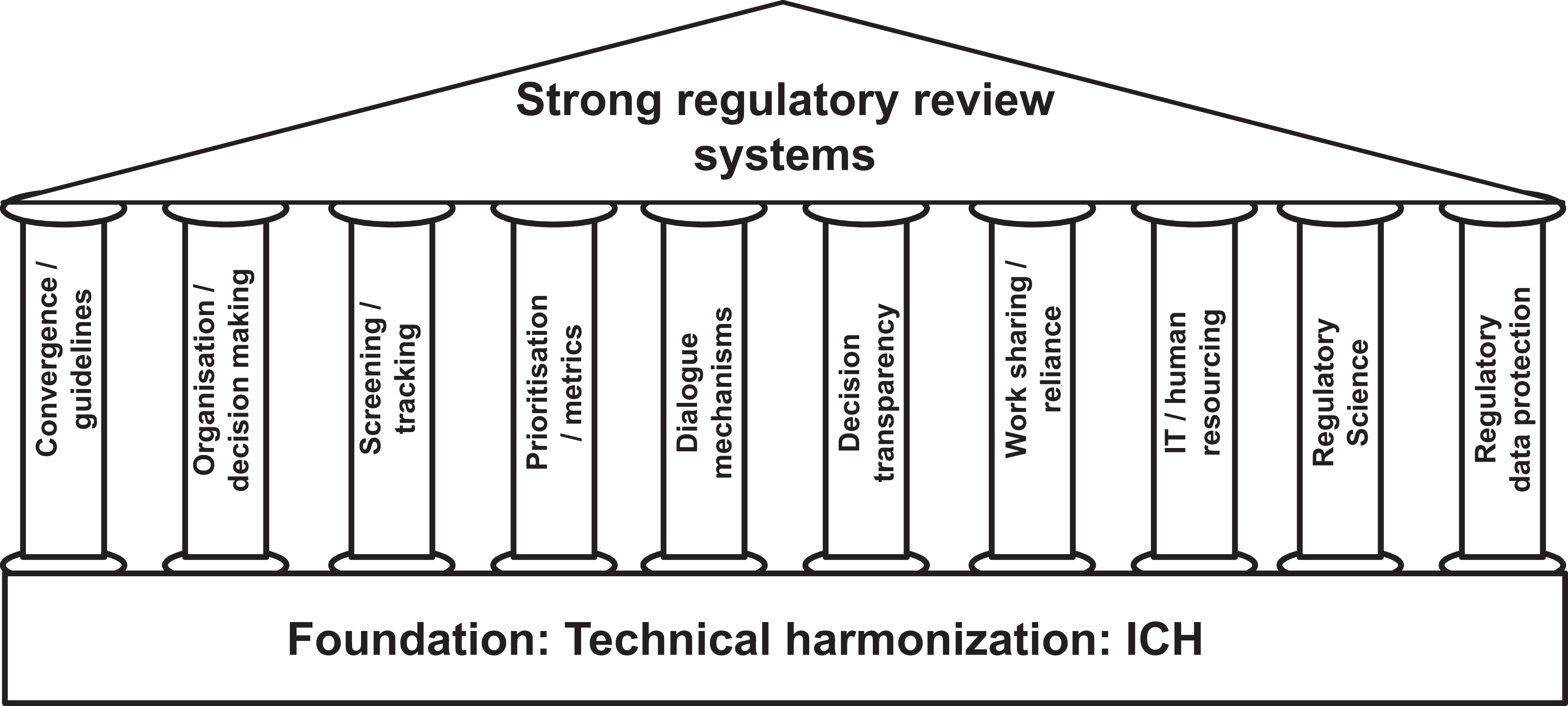
Ten pillars to strengthen regulatory review built on the foundation of technical harmonization.
Ten Pillars That Represent the Key Hallmarks of Strong Regulatory Review Systems
We present our analysis below and invite the perspective of regulators and other stakeholders (The pillars are numbered for ease of reference purposes only).
1. Strong Support for Regulatory Convergence, Guideline Development, and Review
Regulatory convergence is a process whereby regulatory requirements across different countries and regions become more aligned over time. Regulatory convergence does not require formal harmonization of laws and regulations, thus making it a more realistic and achievable goal. 13,14 A good example of the benefits of technical convergence via ICH is the creation of a harmonized approach to adverse drug reaction reporting via the Medical Dictionary for Regulatory Activities (MedDRA) in the 1990s. 15 We believe regulatory authorities should continue to apply and implement ICH guidance where it exists to maximize the benefits of technical harmonization even if they are not members of ICH. Regulatory authorities should also advance convergence efforts beyond technical requirements by adopting appropriate aspects of the 10 pillars presented. Furthermore, we encourage regulators to work together to share information and best practices on general ways of working and exchange ideas on how to maximize efficiencies without compromising applicant/product-specific trade secrets in manufacturing. Taken together, these elements will reduce unnecessary duplication without compromising safety and effectiveness and enhance efficient global development and subsequent approval of medicines.
The need for scientifically driven regulatory requirements is a fundamental principle underpinning regulatory convergence. In its absence, regulatory convergence will flounder as disparate regulatory requirements begin to emerge across different countries and regions. These add time, complexity and cost to the system without adding value from a scientific or patient focused perspective.
In addition, regulatory guidelines, irrespective of whether they are developed at a national or transnational level, play a critical role for both industry and regulatory reviewers, ensuring clear, consistent application of transparent requirements throughout the development and regulatory review process. When developing guidance, regulatory authorities should do so in a clear and transparent manner, engaging in meaningful consultations with a wide range of stakeholders to seek input on draft guidance. This should include reasonable time frames to respond (at least 60 calendar days) and transparency on the suggestions from stakeholders. This 2-way dialogue will result in fit-for-purpose guidance that is future-proofed to allow room for scientific developments. Guidance should be balanced to provide sufficient clarity for most situations while permitting some degree of flexibility, allowing the applicant to diverge from the guidance where there is a scientific justification and agreement with the regulators on an individual program basis. For complex and emerging topics, a good practice is to hold public consultation meetings where all stakeholders may contribute, for example, FDA and EMA public workshops. 16
2. Clear Structure, Organization, and Decision Making
An organizational structure that optimizes the efficient flow of applications through the review process from submission to regulatory decision is essential. There are various ways in which this can be achieved, and the specifics can and should vary depending on the authority’s needs. For example, the US FDA organization of reviewers by therapeutic area produces a strength of deep therapeutic area knowledge. In Europe, strong networks exist whereby national regulatory authority staff may act in a pan–European Union (EU) capacity to review applications for pan-EU decisions. This construct facilitates work sharing and reliance, and a similar model is being developed via the Africa Medicines Authority. 17
Whatever structure is adopted, there is a need for strong reviewer expertise in assessing safety, quality, and efficacy with clear remits of responsibility and justification of decisions based on science, data, and according to transparent and impartial criteria. There should be a possibility to appeal the regulatory authority’s decisions, including ultimately to an independent body (within or outside of the Agency) or court as appropriate. Regulators may elect to use external experts from academia. External experts must have appropriate knowledge, skills, and experience to conduct an assessment; have no conflicts of interest; meet preagreed deadlines; and respect confidentiality of data. Finally, in order to hold applicants to account, the responsibilities of a marketing authorization holder (MAH) need to be clearly defined (including sanctions for noncompliance) in order to safeguard public health on an ongoing basis across the entire product life cycle.
3. Effective Application Screening and Review Tracking Mechanisms
An effective dossier screening phase and process prior to regulatory assessment maximizes regulatory reviewer resources by ensuring that only applications that are complete may proceed through to review and so avoids regulatory authority staff spending time on incomplete applications. It also incentivizes industry to produce high-quality submissions. If minor components are missing, then there should be the opportunity for the applicant to correct this within an agreed short time frame and then allow the review to proceed.
Exceptionally, there should be the possibility for the regulator to require an applicant to re-submit the dossier altogether if the dossier has, for example, been filed prematurely without a major component such as a required clinical study that cannot be generated in a timely fashion. If regulatory agencies are held to account on performance timelines for reviews, then applicants should be equally mindful and ensure that a dossier is materially complete on application and ready for review. By the same token, regulatory agencies should not allow screening failures to be used as a means of managing workload and still meeting metrics. These situations may be avoided by publishing transparent criteria so that the grounds for screening failure are clear to both applicant and regulator.
We suggest that this screening process by the regulator is short (eg, 2-4 weeks), applicable only for major submissions, and is based on criteria relating to common technical dossier components and not non–value added administrative declarations and documents. Ideally the process should be managed electronically.
Once screening is complete and an application proceeds through review then the review phase should have clearly defined milestones whereby the applicant is informed as to how the review is progressing. There should be defined timelines for regulators raising queries and companies responding to them.
4. Commitment to Prioritization and Transparent Metrics
Regulatory agencies are increasingly developing mechanisms to prioritize review of certain medicinal products. We propose that applications should be prioritized on their potential to have the greatest impact to public health issues in the country, irrespective of whether the applicant is a local or multinational company. The opportunity for individual applicant-authority planning meetings for anticipated future submissions (see pillar 5 below) can also help facilitate both prioritization and resource planning by an authority.
Prioritization may involve an expedited review mechanism (ie, shorter review timelines or earlier submission with promising data) for highly innovative drugs according to clearly transparent criteria, for example, pathways exist in US, EU, and Japan. 18 –21
In some markets, there are also mechanisms to prioritize generics or biosimilars of high-priority products, for example, US, South Africa, or Brazil. 22 –24
The concept of “rolling review” to hasten the availability of particularly impactful drugs could also be considered. Generally, a rolling review is applied only in exceptional cases where there is a pressing public health need. The US provides a rolling review system for products with Fast Track or Breakthrough Therapy Designation. 25 Additionally in 2014, the EMA introduced a rolling review system for companies developing vaccines against the Ebola virus. 26
Rolling review involves the agency reviewing portions of an application before the sponsor submits the whole application. On occasions, regulators may consider that a rolling review is merited, but it must be emphasized that this is not appropriate as a standard approach, as regulators may understandably be uncomfortable assessing partial data when resolving a regulatory concern arising during application assessment.
Prioritization can also play a role in addressing postapproval changes, for example, offering expedited review for an urgent safety update or manufacturing change. Adopting a risk-based approach for post approval changes may prevent backlogs in applications and contribute to stronger supply chain resilience globally. 27 Such an approach allows administrative items to be notified without the need for prior approval by the regulatory authority while major changes, which may impact quality, safety, or efficacy of the product, would require preapproval either by the regulators themselves or via the regulator choosing to rely on another leading regulatory authority approval, for example, as outlined in WHO guidances. 28,29
As stated in Table 1, regulatory agencies should have a range of available application procedures (eg, standard, abridged, and fast track). For all these application procedures, authorities should publish review timelines for different types of applications and track progress against these via published metrics.
5. Mechanisms for Applicant-Authority Dialogue Across the Product Lifecycle Should Be in Place
From a pharmaceutical industry perspective, the ability for dialogue with regulators to ensure that your product meets their requirements in advance of filing is critical to avoid spending time and resources on conducting studies that do not support approval. Yet this facility is missing or may even be prohibited altogether in certain low- and middle-income countries (LMICs). The existence of regulatory guidance per se does not always obviate the need for dialogue as uncertainties in the application of the guidance to a specific drug development program may exist or there may be a justification for diverging from the guidance that requires discussion. The form that such dialogue could take varies from the ability to clarify a simple question via email or telephone to more complex topics requiring the submission of a briefing document summarizing the issues and the company’s position with questions requesting advice. This is often needed for situations such as developing a new drug or biologic or for managing a complex postapproval change.
Mechanisms for dialogue should be in place that span the entire life cycle from development through to regulatory review and postapproval and include different types of request (eg, in-person meetings, teleconference discussions, written advice). The option for timely clarification of what is being asked in health authority queries is important. Timelines to respond to queries can be further compressed if the company first needs to translate native language queries to allow subject matter expert input.
In summary, there are clear benefits to both regulators and industry in allowing dialogue with appropriate safeguards. However, we recognize there are potential pitfalls if this is not well executed, with dialogue becoming a resource drain for regulators with no commensurate benefit. Ideally the regulator needs to balance the benefit of meeting with applicants, where this is merited to facilitate efficient drug development against meetings to discuss purely routine issues, which potentially drain resources needed for review without adding value.
Many agencies charge a fee for formal advice requests, and this can be used to resource the process. It is sometimes possible to issue written advice for more straightforward requests, saving resources by reserving in-person meetings for complex and novel problems.
Another potential pitfall that can occur is the requirement for a face-to-face meeting with the applicant for administrative reasons before an application can be accepted for filing. This leads to a bottleneck situation where applications are held in a queue pending the availability of appointment slots. This contrasts with well-executed presubmission meetings to discuss and clarify expectations between applicants and regulators on data to be submitted and its location in the submission. This can be beneficial for regulators as resolution of any points will avoid downstream delays on both sides. Our guiding principle is that meetings between regulators and industry should always be value added for both parties.
6. Transparency on Marketing Authorization Review Decisions
Increasing transparency on marketing authorization review decisions by all regulatory authorities would offer significant benefits to patients, health care professionals, and industry. Publication of an assessment report that summarizes the outcome of marketing authorization reviews for product approvals (and if legally permissible rejections), will both increase visibility of authority decisions and identify publicly which products have a marketing authorization, which is not currently available in all LMICs. These do not need to contain all the detail available in FDA or EMA reports; instead, these reviews could follow a standardized outline that includes as a minimum the basic elements assessed to demonstrate that the authority is applying consistent standards to the review in order to enhance public confidence and trust. The International Pharmaceutical Regulators Forum (IPRF) model for biosimilars is a good exemplar of one such template. 30 However, proprietary and/or commercially confidential information should be redacted and not be included in any public assessment report. The applicant should have the opportunity to review any assessment report produced and propose redactions of such confidential information prior to publication to ensure this.
Publication of the rationale for approvals enables applicants to understand more fully what is required for MAAs in each country. This should improve the standard and consistency of incoming applications, thereby enhancing efficiency for both regulator and industry. Publication should also occur in a predefined and reasonable time frame to enable all stakeholders including health care professionals and patients to access the information. This will also be important to support reliance mechanisms (see pillar 7 below).
7. Commitment to Work-Sharing, Training, Recognition, and Reliance
There is increasing international focus on work-sharing, reliance, and recognition as methods to both address capacity gaps within regulatory authorities and to strengthen regulatory expertise. The WHO’s Global Benchmarking Tool and Good Regulatory Practice documents both anticipate that where appropriate/applicable a regulatory authority should engage in work sharing to share resources and build capacity and expertise. 31,32 This is also promoted by nongovernmental organizations such as the Bill and Melinda Gates Foundation (BMGF) as a way to strengthen regulatory systems in LMICs. 33
However, we emphasize that recognition and reliance are not processes that are only undertaken by regulatory authorities in LMICs. Increasingly, we see use of these processes even by leading regulatory authorities. A recent example is the ACSS Consortium (a collaborative initiative of medium-sized regulatory authorities from Australia, Canada, Singapore, and Switzerland) who undertook a Generic Medicines Work Sharing Trial for the coordinated assessment of a generic application filed with multiple ACSS Consortium agencies. 34,35 Recognition is also undertaken to reduce the burden of duplicative inspections by multiple leading regulatory authorities. For example, the EMA has Mutual Recognition Agreements (MRAs) with non-EU/EEA authorities (now including the US FDA) concerning Good Manufacturing Practice (GMP) inspections. 36 Uptake of schemes such as Pharmaceutical Inspection Co-operation Scheme (PIC/S) are also increasingly being utilized by regulatory authorities to harmonize GMP standards and inspections and reduce the burden on regulatory authorities. 37 Recognition of Good Clinical Practice (GCP) and GMP inspections from other countries/regions without the need to duplicate these inspections, especially those conducted by US FDA and EMA or other leading regulatory authorities, would simultaneously reduce delays in the review process and increase capacity within regulatory authorities.
Another example where non–value added requirements may cause delays is additional duplicative local batch release testing of samples by regulatory agencies as a prerequisite for regulatory approval. This generally does not add value to the review process and should be avoided; however, this is separate from the legitimate need to test products to detect falsified medicines in the supply chain.
Recognition of batch testing results, marketing authorizations, and postapproval changes from other regions with leading regulatory authorities should also be considered. Efficient reliance practices could be applied such that the authority focuses their review resources on aspects that may need to be determined nationally (eg, labeling) and could rely in whole or in part on evaluations done by other leading regulatory authorities while respecting their own sovereignty for ultimate decision making. 31,32 This could reduce backlogs in applications exacerbated by postapproval changes. Publication of the rationale for approvals enables applicants to understand more fully what is required for Marketing Authorization Applications (MAAs) within each country. However, reliance will also depend on either publication or sharing of assessment reports within a pre-defined and reasonable timeframe (see pillar 6 above).
8. Supportive Information Technology (IT) Infrastructure and Human Resourcing
Advancements in IT have improved scientific innovation and clinical development to the benefit of public health. IT systems help facilitate regulatory research and the advancement of regulatory science (see pillar 9 below). 38 Robust IT infrastructure will help regulatory authorities increase their capacity and ability to perform faster reviews, and to better manage changes in anticipated workload. For example, acceptance of eCTD format submissions can help optimize resources, drive efficiencies and facilitate tracking of applications during review, and ensure the content of submissions are consistent with global standards. Regulatory authorities are encouraged to partner with industry in adopting new technologies by running pilots or other engagements that would be beneficial for regulatory authority staff. IT systems can be leveraged to check in and track applications throughout regulatory review and also provide a means to assess current and projected workloads, enabling the regulator to plan ahead. State-of-the-art video and teleconferencing tools will facilitate regulators’ collaboration with each other and also with applicants.
Regulatory authorities must also be appropriately funded and resourced and be empowered to recruit and train staff or engage with academic experts. If regulators do not have oversight of a ring-fenced budget to finance their activities then this can act as a barrier to introducing any of the features in this publication as there will not be assigned resourcing and funding.
9. Commitment to Advancing Regulatory Science
Regulatory science can help regulatory authorities build and develop scientific capacity to anticipate future needs and engineer new paradigms to address scientific advances and thus to develop new and more effective approaches to regulatory review. 39,40 Factors likely to impact drug development in the near future include the genomic revolution, the rise in targeted therapies or personalized medicines, the availability of digital health data, the rising importance of real world evidence in decision making, and advanced therapies and complex drug-device and novel drug-drug combinations to name a few. 41 Regulators should aspire to stay up-to-date with advancing regulatory science as a means of tracking future demand versus capacity and expertise so that resourcing and staff development and education bottlenecks do not impede the timely review of new medicines and/or complex generics and biosimilars. Continuous development of tools and systems are also needed for comprehensive monitoring of marketed products to safeguard public health. Robust and contemporary IT systems, as described in the section above, also facilitate regulatory research and advancement of regulatory science, ensuring that regulatory authorities can leverage the full remit of their insights and state-of-the-art knowledge. In the light of such advances, it becomes critical for regulators to share their experiences with each other (see pillar 1 above) as scientific advances stimulate new challenges and opportunities.
10. Support for Innovation via Regulatory Data Protection
All regulatory agencies should support innovation and the research and development of new medicinal products by providing a reasonable period of regulatory data protection for the test and trial data submitted by original applicants in support of their MAAs, in accordance with TRIPS Article 39.3. 42,43 During this time, the innovator’s data file may not be relied on by other applicants to support competitor or copy applications.
Next Steps: Challenge/Call for Action
It is in the interest of all stakeholders to have effective and efficient regulatory review systems in place. From development and registration of new, innovative products for unmet medical need to the management of approved products through their life cycle, there is a pressing need to ensure streamlined regulatory review systems that result in safe and effective medicines for patients. As outlined in this paper, many decades of work have resulted in positive strides toward this goal across all jurisdictions, most notably in the well-established pharmaceutical markets (eg, US, EU, Canada, Japan, Switzerland, and Australia). Today, there are multiple efforts in progress to support this goal globally including those driven by regulatory authorities, WHO and its regional offices, APEC, etc, and industry associations such as IFPMA, EFPIA, FIFARMA, and PhRMA 8 ; however, notwithstanding this progress, gaps in regulatory capacity and capability continue to exist, especially among regulatory authorities in LMICs. In this paper, we distil what we believe to be the key framework elements that contribute to good regulatory review systems to realistic, actionable goals. We believe progress across these 10 areas would have a positive impact on regulatory review systems to the benefit of all stakeholders. With respect to individual regulatory authorities, it will be the case that not all of these areas for action will be relevant at any given point in time, and the degree to which they are adopted will need to fit the specific national agency’s size, level of resourcing, and local priorities. Thus, we would encourage regulatory authorities to consider which of the areas might be of most benefit to their particular situation and what particular actions/initiatives would be useful. It is possible that implementing even simple basic measures could have significant benefit on the overall system, for example, establishment of an effective application screening process at the regulatory authority.
While all regulatory authorities should have appropriate mechanisms for strong regulatory review, it must be acknowledged that not all regulatory authorities will be, or should strive to be, leading regulatory authorities as discussed by the WHO’s draft Good Regulatory Practices: Guidelines for National Regulatory Authorities for Medical Products. 32 To this end, international regulatory cooperation, for example, reliance mechanisms between regulatory authorities, should be encouraged and expanded where appropriate. The successful use of such mechanisms by leading regulatory authorities reminds us that reliance is an important part of the overall system and is for all regulatory authorities whether small or large.
This commentary is intended to highlight areas that could be considered for action within regulatory authorities. We acknowledge that there are several potential limitations with these proposals. First, this is the view of one multinational company and hence may not be generalizable to other multinational companies with different research focus and business priorities. Second, smaller companies may have a different perspective if for example they specialize in fewer markets, or specific classes or types of products where they may have experienced different pain points in reviews to those that we have observed. Finally and perhaps most importantly, the regulatory authorities themselves may have differing views on whether these are the most appropriate 10 items to be prioritized. We did not seek review by regulators in the development of this list at this stage as our aim was to share the perspective of a multinational company. However, consultation with them could be considered as a follow-up.
Our hope is that by writing this article we may stimulate discussion and debate in this area among relevant stakeholders. As part of the overall health care ecosystem, we believe dialogue between regulatory authorities and key stakeholders, such as industry, patient groups, governments, WHO, and other stakeholders, for example, BMGF could be an important follow-on step to build momentum in this area with the aim of benefiting patients irrespective of where they live. We therefore invite all interested parties to engage in a dialogue on the features identified in this paper and in this area more broadly.
Footnotes
Acknowledgments
We acknowledge the review, advice, and suggestions received from Pfizer regulatory colleagues around the world.
Declaration of Conflicting Interests
The authors are employees and shareholders of Pfizer and work in regulatory policy.
Funding
No financial support of the research, authorship, and/or publication of this article was declared.