
Abstract
The Health and Environmental Sciences Institute (HESI) Developmental and Reproductive Toxicology Technical Committee sponsored a pharmaceutical industry survey on current industry practices for contraception use during clinical trials. The objectives of the survey were to improve our understanding of the current industry practices for contraception requirements in clinical trials, the governance processes set up to promote consistency and/or compliance with contraception requirements, and the effectiveness of current contraception practices in preventing pregnancies during clinical trials. Opportunities for improvements in current practices were also considered. The survey results from 12 pharmaceutical companies identified significant variability among companies with regard to contraception practices and governance during clinical trials. This variability was due primarily to differences in definitions, areas of scientific uncertainty or misunderstanding, and differences in company approaches to enrollment in clinical trials. The survey also revealed that few companies collected data in a manner that would allow a retrospective understanding of the reasons for failure of birth control during clinical trials. In this article, suggestions are made for topics where regulatory guidance or scientific publications could facilitate best practice. These include provisions for a pragmatic definition of women of childbearing potential, guidance on how animal data can influence the requirements for male and female birth control, evidence-based guidance on birth control and pregnancy testing regimes suitable for low- and high-risk situations, plus practical methods to ascertain the risk of drug-drug interactions with hormonal contraceptives.
Introduction
Background
There are no specific, harmonized, international regulatory guidelines on birth control requirements and pregnancy prevention within clinical trials. Although the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) provides some recommendations in M3(R2) 1 regarding circumstances in which “highly effective” methods should be used, the guidance document does not suggest what methods might achieve the desired level of “high” efficacy. Some individual countries provide more detailed guidance to birth control. Interpretation and implementation of (for example) the UK Medicines and Healthcare Products Regulatory Agency (MHRA) Contraception Guidelines 2 for clinical trials has highlighted misunderstandings in the biopharmaceutical industry and in the regulatory community regarding the relative effectiveness of these contraceptive methods and the role of preclinical information in influencing the choice and duration of use of birth control for women and men in clinical trials. Given the lack of overarching international regulation, and the variability across biopharmaceutical companies regarding common birth control methods, it is not surprising that industry approaches to preventing inadvertent pregnancy exposure in clinical trials have been shown to vary quite considerably. 3 This lack of consistent guidance has caused some uncertainty in clinical protocol content leading to requests for country-specific amendments to what were intended to be globally consistent protocols and informed consent. Recent guidance from the Clinical Trial Facilitation Group (CTFG) of the European Heads of Medicines Agencies (HMA) 4 has helped provide a harmonized European view on these topics that was not available at the time this industry survey was carried out.
Objectives
The International Life Sciences Institute–Health and Environmental Sciences Institute (HESI) Developmental and Reproductive Toxicology (DART) Technical Committee formed a Birth Control Working Group to explore these issues. It undertook an informal survey of member companies with the objectives of improving our understanding of (1) the current industry practices for contraception requirements in clinical trials for both women and men, (2) the governance processes set up to promote consistency and/or compliance with contraception requirements, and (3) the effectiveness of current contraception practices in preventing pregnancies during clinical trials. This study presents the results of this industry survey and identifies opportunities for improvements in current practices.
Methods
The survey covered 5 distinct themes that included questions pertaining to management of women of childbearing potential (WCBP) and male participants in clinical trials. The survey explored the governance processes set up to promote consistency and/or compliance with contraception requirements, the methods used to determine risk of drug-drug interaction effects on hormonal methods of contraception and, if available, information on the effectiveness of current contraception practices in preventing pregnancies during clinical trials. These 5 themes (parts A to E) are listed in Table 1.
Survey themes.

From the outset, it was appreciated that because of the divergence in the content of the 5 themes, the responses from companies would likely require contributions from different specialists within the global pharmaceutical company. It was also realized that some companies may not be able to answer all 5 elements because the relevant information or specialist could not be identified. Therefore, partially completed surveys were accepted. The survey was also designed to assist the authoring team to discuss and develop areas for improvement within their own companies in their approach to this interdisciplinary topic.
This survey was distributed via a single company representative to the 15 HESI DART participant biopharmaceutical companies. In addition, 2 other pharmaceutical companies were sent the survey following requests to participate. A total of 12 responses (10 HESI DART companies, 2 additional companies external to HESI DART) were received from companies headquartered in United States, Europe, and Japan with significant differences in size as well as disease and modality-focus. The responses were anonymized before review by the authoring team. For each of the 5 themes, the survey questions and the responses are summarized in text or tables followed by comments from the authoring team that are specific to the individual themes. The final discussion brings together the interlinked issues and identifies areas where the authoring team believes harmonized approaches, preferably underpinned by international guidance, would be particularly helpful.
Results and Comments
Part A: Company Guidance for Contraception and/or Barrier Protection for Male Participants in Clinical Trials
1. Is there company guidance for contraception or barrier protection requirements for men in clinical trials?
Summary of key factors considered by respondents with requirements for contraception and/or barrier protection in male subjects.

2. If contraception or barrier protection use is specified, how is the duration of posttrial contraception determined?
Duration of posttrial contraception.

3. Is double-barrier protection ever required? If yes, under what circumstances?
Circumstances where double-barrier protection is required.

4. Are 2 forms of highly effective contraception ever required? If yes, under what circumstances?
5. Are there company restrictions or guidance for enrolling men with pregnant partners? (yes/no) If yes, please describe.
6. Do contraception and barrier protection requirements differ between small molecule and biologic drugs?
Part A Commentary
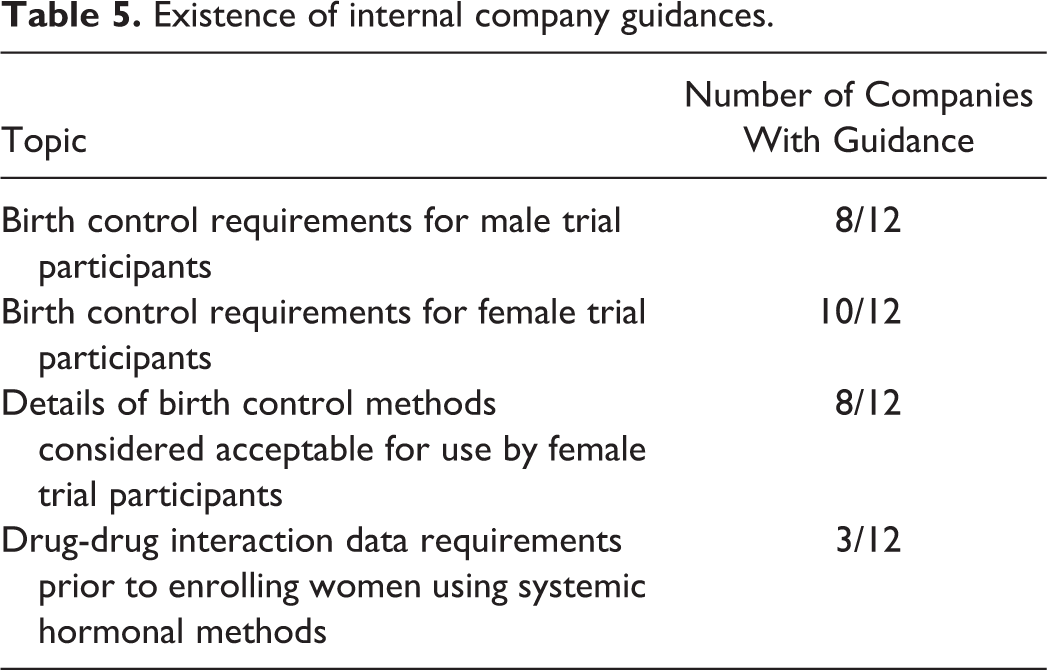
One-third of responding companies did not have (or could not locate) internal guidances on contraception for male participants in clinical trials, which could in itself be a source of inconsistency between projects regarding the decisions reached.
Broadly speaking, the need for male participants to use birth control could arise from 3 different concerns: first, concerns regarding the quantity or quality of spermatozoa such that time to conception or chances of having a very early pregnancy loss in a female partner may increase; second, concerns regarding excretion of the drug in seminal fluid that might adversely affect the safety of the sexual partner (eg, male-mediated transfer of a genotoxin); and third, concerns regarding excretion of the drug in seminal fluid that might adversely affect the outcome of an existing pregnancy. All of these concerns could potentially be mitigated by use of male condom by the male healthy volunteer or patient. Some responses mention requirement for highly effective methods (<1% failure rate per year) yet, with exception of vasectomy, 5 there are no male methods of birth control that can realistically achieve that degree of efficacy. In those companies that required highly effective methods, it was unclear whether a male participant whose female partner did not (or could not) use highly effective methods would be excluded from trial participation because of their partners’ inability to comply, or whether the male participant could choose to use a barrier method.
For most clinical trials, there is testicular histopathology information from repeat-dose toxicity studies in 2 species to gain insights on presence or absence of effects on spermatogenesis. However, for early clinical trials, the assessment of functional effects on male fertility has not yet been undertaken. In the absence of functional male fertility data, it remains unclear whether and for how long men should be advised not to procreate following participation in a clinical trial. It became apparent during discussions that in this situation, some but not all companies, may advise male participants “not to procreate.” With at least 1 company, the male participant is allowed to choose whether that advice is implemented through using a condom or through birth control methods used by the female partner. It was perhaps unsurprising that there was lack of consistency on the duration of male contraceptive use in that even within those limited regulatory guidances 2 that were available to sponsors, there is reference to either continuing contraceptive use for 5 half-lives or for the duration of a spermatogenic cycle, without provision of clear criteria for choosing one or the other. Some companies clearly linked the duration of condom use with lack of preclinical data on the effects of the compound on embryo-fetal development and/or evidence that the compound can cause fetal harm when directly administered to a pregnant animal. The concern for potential embryo-fetal harm arising following vaginal absorption of drugs in the seminal fluid has been explored in a series of experiments sponsored by the HESI DART group that are reported elsewhere. 6 –10
Part B: Company Guidance on Management of WCBP in Clinical Trials
1. Is there a company-wide definition of a WCBP? Please provide WCBP definition.
Permanent cessation of menstruation for at least 12 months prior to screening in women who are 45 years of age or older. Permanent cessation of menstruation for 2 years. Amenorrheic for at least 12 months or amenorrheic for at least 6 months and serum follicle stimulating hormone (FSH) concentrations of >40 mIU/mL. At least 1 year since last regular menses with an FSH >40 IU/L or at least 5 years since last regular menses 12 months of amenorrhea in a woman over age 45 years in the absence of other biological or physiological causes, plus women under the age of 62 years must have a serum FSH level >40 mIU/mL. 12 months with no menses without an alternative medical cause or no menses for at least 2 years. As over the age of 60 years, or between 45 and 60 years being amenorrheic for at least 2 years with plasma FSH level >30 IU/L.
In addition, although women who have undergone a hysterectomy or bilateral salpingo-oophorectomy were consistently considered surgically sterile (and, therefore, not of childbearing potential), there was inconsistency in whether a woman with bilateral tubal ligation was considered not of childbearing potential.
2. Is there company guidance on birth control requirements for WCBP in clinical trials? If yes, please describe the basic requirements and governance process.
Existence of internal company guidances.
3. If contraception or barrier protection use is specified, how is the duration of posttrial contraception determined? Please describe.
One company varies its posttreatment contraception requirements based on the pharmacokinetics data of the compound. The remaining company uses an algorithm that incorporates hazard and exposure information, wherein if adverse embryo-fetal effects are observed in nonclinical studies, the duration of contraception postdosing is the time it takes the plasma drug level to drop below a safe level (NOAEL or NOAEL divided by an appropriate safety factor for findings of teratogenicity). However, if no embryo-fetal data are available, the duration of contraception postdosing is “the time it takes the plasma drug level to drop below the NOEL for pharmacology.”
4. Is there company guidance that details the birth control methods considered acceptable for use? eg, male condom, combined oral contraceptives (COCs), copper-banded intrauterine devices (IUDs), etc. Please provide a list of recommended methods.
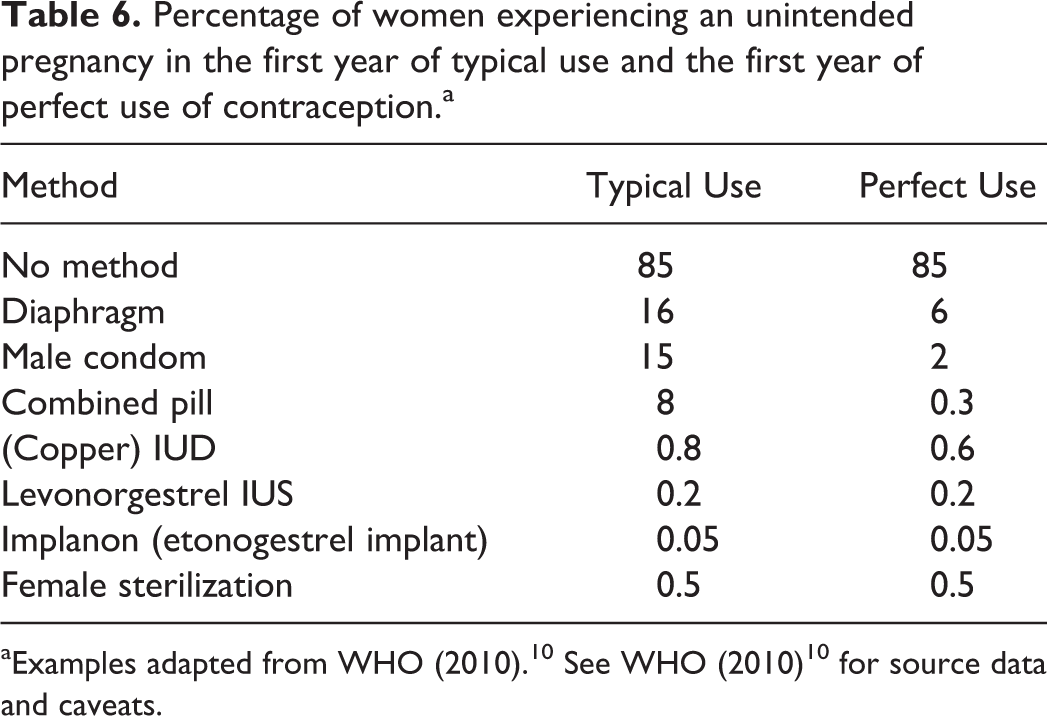
Percentage of women experiencing an unintended pregnancy in the first year of typical use and the first year of perfect use of contraception.a
Part B Commentary
Seven different definitions of postmenopausal were elicited by the survey. Although all definitions mention cessation of menstruation, the duration of that cessation varied from 6 months to 5 years. Three definitions mentioned specific ages, all of which were applied differently. Four of those definitions required measuring FSH with verification of a value greater than either 30 or 40 IU/L. It is unclear if this variation arises from deliberate desire to be more stringent in particular circumstances or whether it has simply arisen through company custom and practice. None of the companies used the definition provided in ICH M3(R2), which is “postmenopausal is defined as 12 months with no menses without an alternative medical cause.”
Many companies considered that a woman with a bilateral tubal ligation (or occlusions) to be “surgically sterilized” and “not of childbearing potential.” However, according to data quoted by the WHO (with examples shown in Table 6), the method failure rate of bilateral tubal ligation can be higher than some modern long-acting reversible hormonal birth control methods. 5 It is understood that failure rates following tubal ligation or occlusion procedures can vary with age of the women at time of procedure and exact surgical method employed. 11 In a large, global phase III clinical trial, where neither the patient age at time of the tubal procedure nor the exact surgical method employed can be controlled, it seems potentially problematic to assume tubally ligated women are incapable of conceiving.
ICH M3(R2) 1 states, “highly effective methods of birth control are defined as those, alone or in combination, that result in a low failure rate (ie, less than 1% per year) when used consistently and correctly” but offers no guidance on what those methods might be. The previous version of ICH M3 offered the additional information that “implants, injectables, combined oral contraceptives, some IUDs, sexual abstinence or vasectomized partner” were highly effective, which concurs with the failure rate data in WHO guidance. 5 However, although most companies gave instruction on whether or not a highly effective method is required, few provided details of what those highly effective method might be, which leaves the health care provider and/or patient to determine their best options. A minority of surveyed companies provided guidance to WCBP or their clinical trial–participating partner on methods that were considered “highly effective” that approximated those described previously in ICH M3 or by WHO, and are now described in the CTFG 2014 guidance document. 4
Part C: Influence of Animal Data on Birth Control Requirements on Clinical Trials
1. Is there policy/guidance on what developmental toxicity animal data should be available before WCBP are enrolled in clinical trials? If yes, is the guidance different for small molecules versus biologics?
A majority of companies with policies require the results of preliminary embryo-fetal development (EFD) studies in 2 species before enrollment of WCBP in clinical trials of limited scope and duration, as specified in ICH M3(R2) for small molecules. Interestingly, one company indicated that it will proceed into clinical trials of limited scope and duration with WCBP based on preliminary data from one species for some anti-infective programs where there is no mammalian target. Two respondents indicated they follow ICH S9 for advanced cancer indications and, consistent with that guidance, no pregnant animal work would be performed prior to the conduct of clinical trials in WCBP.
For biologics, the evaluation of reproductive toxicity is only conducted in pharmacologically relevant species. When the only relevant species is a nonhuman primate, many companies now use an enhanced pre-/postnatal development (e-PPND) study design as suggested as an option in ICH S6(R1)
12
to maximize the information obtained. For monoclonal antibodies for which embryo-fetal exposure during organogenesis is understood to be low in humans, consistent with ICH M3(R2) and ICH S6(R1), the e-PPND study is often conducted during phase III, with the results available for submission of the marketing application.
2. When/if proceeding into WCBP clinical trials without EFD studies in 2 species, what difference (if any) does that make to birth control or pregnancy testing on the trial?
For the companies that indicated the absence of EFD study results would not make any difference in birth control or pregnancy testing requirements, they responded that they generally employ stringent requirements in these areas during clinical development.
For biologics, a case-by-case approach is followed by some companies, including conducting target liability assessments. Some companies reported that for targets considered a high risk for causing fetal harm, the e-PPND study may be accelerated and/or a male condom could be added to the primary method employed by the female participant, while less restrictive birth control requirements (eg, a single effective method) would be specified for targets not predicted to be associated with adverse fetal development. Some companies would not require a second method of contraception for biologics regardless of the target liability for embryo-fetal harm.
3. For WCBP on trials, when animal data illustrate potential for fetal harm at clinical exposures, what difference (if any) does that make to birth control or pregnancy testing on the trial?
For companies responding that positive EFD results would make no difference, they indicated that the same (strict) procedures are enforced for WCBP enrolled in clinical trials regardless of the nonclinical results. One respondent indicated that progression in clinical trials with a probable developmental toxicant would be highly dependent on the indication (risk/benefit ratio).
4. For men on trials, when animal data illustrate potential for fetal harm at clinical exposures, what difference (if any) does that make to birth control requirements on the trial?
For those companies responding that positive EFD findings would make no difference for birth control requirements for men, the default procedures were considered stringent enough. Another company reported that such results would have historically had no effect unless required by a regulatory agency, but they recently recommended the use of contraception for men on clinical trials based on the EFD results.
5. For men on trials, when animal data illustrate potential for testicular toxicity at clinical exposures, what difference does that make to informed consent and birth control requirements on the trial?
An alternative approach from one company was to advise the male participant “not to try to father children during the trial and for a period afterwards” (generally for 3-6 months), with the means of avoiding procreation left to the choice of the participant.
Some companies indicated that development of a testicular toxicant would be unlikely unless the benefit outweighed the risk (eg, oncology, geriatric populations), reversibility of the effect was demonstrated, or the finding was shown not to be relevant to humans.
6. When the embryo-fetal and male and female fertility data indicate no risk, does this information impact contraception requirements for men or WCBP?
For those companies responding that this information would have no effect on contraception requirements, most indicated that WCBP are required to use an effective method of contraception regardless of the outcome of the animal studies.
7. Does the margin of safety (MOS) for embryo-fetal toxicity impact contraception requirements?
For companies responding that MOS did not affect contraception requirements, the most conservative approach is taken for WCBP in clinical trials. It was also noted by one company that WCBP are required to use contraception independent of MOS considerations since pregnancy is considered a discontinuation criterion.
8. Other comments
Part C Commentary
Not surprisingly, all companies were, in effect, following ICH M3(R2) 1 regarding the animal work required to support clinical trials, and the majority (but not all) were utilizing the flexibility of the revised guideline to proceed into clinical trials with WCBP on the basis of preliminary assessment of embryo-fetal toxicity rather than waiting for the full regulatory EFD studies. It was noted that consistent with ICH M3(R2), biologics would often proceed into clinical trials in WCBP without nonclinical EFD data. In such a situation, some but not all companies adjust their clinical trial management procedures. This difference may simply reflect the variation with which companies approach the use of birth control methods that have either perfect use failure rates or real-world failure rates that meet the ICH M3(R2) intended desire of <1% per year.
Part D: Liability of DDI of the Investigational Drug With Systemic Hormonal Contraceptives
1. Is there company guidance on the DDI testing required prior to allowing use of systemic hormonal contraceptives?
“Projects are expected to undertake a specific testing cascade prior to enrolling WCBP using hormonal methods.” “A drug-drug interaction study must demonstrate that the effectiveness of the hormone based contraceptive has not been adversely affected by the investigational drug. Or there must be compelling evidence to substantiate that the investigational product(s) or concomitant medications will not adversely affect contraception effectiveness.” “If the drug has been identified as a potential inducer of cytochrome P450 (CYP)3A4 based on in vitro screening, then reliance on hormonal contraceptives as a means of effective contraception is not allowed in clinical trials until after completion of a pertinent drug interaction study that rules out CYP3A4 induction.”
From these responses, it would appear that having DDI information is a fundamental and important component of their decision-making process. The remainder (8/12) indicated either no guidance or decisions were made on a case-by-basis basis (1 company did not respond to this particular question).
2. What default testing for DDI is required?
3. Would a final submission package generally include a DDI study with ethinyl estradiol/COC?
4. What criteria are used to determine if a DDI signal is of sufficient magnitude to be of concern regards either COC efficacy (decreased exposure to estrogen component) or COC/hormone replacement therapy safety (eg, increased exposure to estrogen component)
Part D Commentary
In comparison to the other parts of the survey, this was an area where (written) company guidance was often lacking or was at least inaccessible to the survey respondent. Given that the survey was distributed primarily via the reproductive toxicology representative, the inability to describe company guidance could reflect the fact that the scientific specialists in DDI would tend to reside in separate groups.
However, considering the preponderance of hormonal methods (in many forms including oral, parenteral, and intrauterine devices) as the primary method of choice for many women, it is noteworthy that there is inconsistent guidance across companies on this issue. It is unclear if the more generic guidance on DDI approaches for other common co-medications of the intended patient population is appropriate (in content and timing) to specifically address the DDI questions required to support women enrolling in a clinical trial who wish to continue to use their preferred hormonal contraceptives. In some circumstances, to better understand the risk of birth control method failure, further investigations may be warranted. The literature lacks a current user-led, evidence-based white paper on this specific topic.
Part E: Reporting and Review of Pregnancies in Phase I–Phase III Clinical Trials
1. Is there company guidance on reporting and follow-up on inadvertent pregnancy drug exposure in WCBP in clinical trials? What is the reporting mechanism?
2. In clinical trials where the men were requested to use contraception, is there company guidance on reporting and follow-up of pregnancies in WCBP partners? What is the reporting mechanism?
3. Reported pregnancies—What information is gathered about the (failed) birth control method? What other information is typically gathered about the exposed pregnant patient (eg, primagravida etc).
4. Review of pregnancy rates in trials including WCBP: (a) Is there a routine review of reported pregnancy rates at the end of trials? (b) Is there a review of reported pregnancy rates at the end of trials with test agents considered likely to cause fetal harm? (c) What (if any) oversight of the pregnancy rates across multiple trials is undertaken (eg, internal ethical review board on an annual basis?), (d) Is there a review of pregnancy rates with drugs that have a DDI with hormonal contraceptives? (e) Are pregnancy rates summarized by geographical area or country to understand the cultural impact on contraception practices and effectiveness?
Part E Commentary
Although every company appeared to have explicit guidance on how to report pregnancies in clinical trials, it was apparent that few companies collected data in a manner that would allow retrospective understanding of the reasons for failure of birth control within the clinical trial situation. The survey revealed there was no enhanced interrogation of clinical trials with putative developmental toxicants and no systematic collation of pregnancy rate data. There would appear to be limited opportunity for corporate learning or cross-project learning.
Discussion
Overall Quality and Quantity of Information in Responses
This was a small survey with only 12 responding companies that were predominantly major biopharmaceutical companies. These companies sponsor clinical trials in many countries using different operating frameworks (eg, in-house clinical trials, contract research trials, externally sponsored collaborative trials, etc). Given the size, scope, and global reach of the responding companies, despite the small sample size, the responses are likely to reflect common practices within the biopharmaceutical industry.
The survey confirms that with the exception of provision of drug-drug interaction guidance, the majority of companies have policies or guidances covering the topics included in this survey (Table 5). Of note is that this survey includes biopharmaceutical companies that did not contribute to the Ng et al 3 compilation, yet confirms the variable practices previously flagged by Ng et al.
This survey enabled a more detailed assessment of company procedures than previous surveys and deliberately permitted free text answers. The author group had assisted in providing the information from their own companies—an activity that in itself benefited the participating companies by revealing potential gaps or inconsistencies in knowledge and/or policy. This also meant that the author group could discuss and explore the free text responses from an informed position. The discussion among the author group revealed that companies shared the same intentions (such as minimizing the chances of adverse pregnancy outcome resulting from participation in a clinical trial) and that much of the variability in current practices arose from 3 main issues: first, differences in definitions; second, differences in scientific thinking and lastly, differences in company approaches to enrollment in clinical trials.
Differences in Terminology and/or Definitions
The survey revealed various areas where there were differences related either to definitions or language. Some of these areas of differences have the potential to lead to unintentional adoption of suboptimal and/or cumbersome clinical trial management practices. Some of these are discussed below.
Definition of “postmenopausal”
There was considerable variation in the definition of “postmenopausal”—several of which required blood sampling for FSH concentrations. ICH M3(R2) provides a very simple definition of “postmenopausal”: “12 months with no menses without an alternative medical cause” and although some companies may considered that to be oversimplistic, there could be benefit in agreeing on a definition that reduces the requirement for what may be unnecessary FSH monitoring of female trial participants.
Tubal ligation/occlusion techniques—effective birth control or “sterilization”?
Some companies consider women who have undergone bilateral occlusive or ligation techniques as not of childbearing potential, which would then mean that those women are not monitored for pregnancy. Other companies classify tubal ligation as a highly effective method of birth control and still manage the women as if they were of childbearing potential (eg, would conduct pregnancy test as required by the clinical protocol for any WCBP).
Double-barrier methods
Discussions among the authors revealed a diversity of definitions for this term. Some took it to mean 2 methods, each of which was a physical barrier to sperm (eg, a condom plus cervical cap). Others took it to also include 1 physical barrier method plus the use of a chemical barrier, for example, male condom plus spermicide. Others interpreted it to mean 2 methods, one of which was a barrier. It is not known what definitions were used by the survey respondents. Regulatory guidance from the UK MHRA 2 defines a barrier method as a contraceptive device that physically prevents sperm from entering the endometrial cavity and fallopian tubes (eg, male condom, female condom, or diaphragm). Regardless of the definition that was used by the respondents, at least 5 companies claim “double barrier” as standard and that at least 1 company stated that evidence of potential or known teratogenicity would be a trigger for requiring double-barrier methods. This is of potential concern because, based on expert overview, 5 even in perfect use, barrier methods cannot achieve the efficacy of many nonbarrier methods. Barrier methods were not included in the list of highly effective methods quoted in the previous version of ICH M3 nor in the recent CTFG guidance document. 4 In addition, for those participants who would routinely use a barrier method as their preferred primary method, the requirement to add a second barrier method could actually put them at greater risk of method failure if the second method compromises the first. This was recognized by the MHRA who revised their advice in 2010 2 on double-barrier methods by stating, “A female condom and a male condom should not be used together as friction between the two can result in either product failing” and is reiterated in the CTFG guidance. 4
Description of (highly) effective methods
Many companies do not give advice on which birth control methods are effective versus highly effective (ie, have failure rates of <1% when in perfect use). Birth control methods have variable “typical use” efficacy that is described in international guidelines. 5,14 This lack of specific guidance could leave women vulnerable to choosing a method for a clinical trial that is not the most appropriate for their circumstance. For example, for those companies suggesting that a “double barrier” is a preferred option, they may be using that terminology thinking it will be highly effective, but could unwittingly place a woman at risk of an unplanned pregnancy when she switches from a highly effective hormonal method to less effective physical methods.
Areas of Scientific Differences
Requirement for male barrier methods to avoid seminal exposure of WCBP partner
The survey revealed that some companies would request men to use condoms (or for both partners to use effective contraception) where the male participant was being given a drug that was likely to cause embryo-fetal harm. Discussions revealed these precautions were often put in place to avoid the theoretical risk arising from vaginal absorption of the drug delivered in the ejaculate. However, based on known human seminal fluid concentrations, the risk arising from drug exposure in the ejaculate would appear to be very small. 15 HESI DART companies have undertaken a series of experiments to explore this risk and the outcome of this work has recently been published. 6 –10 These experiments provide further reassurance that the risk arising from seminal exposure of monoclonal antibodies is negligible 8,9 and, even with potent small molecule teratogens, the margin of safety following vaginal absorption is extremely large. 7 During discussion of the different approaches, the authoring team concluded that the reference to continuation of birth control for 5 half-lives after the end of a clinical trial as a “washout” period, although mentioned in regulatory guidance, 4 is probably scientifically unnecessary for male participants who have received nongenotoxic pharmaceuticals. The limited excretion of the pharmaceutic into a small volume of seminal fluid with incomplete vaginal absorption means adverse maternal or fetal effects are unlikely in most clinical trial scenarios.
Requirement for birth control during and following trials in men for reasons related to male reproductive hazard
Most early clinical trials with small molecules proceed with information on genotoxicity and on male reproductive tract histopathology from repeat dose toxicity studies, but in the absence of the (rodent) mating studies that holistically assess male fertility. In this circumstance, how companies approach advising male participants was an area of considerable diversity, where some companies adopted default requirements for condom use for several months (ie, emulating the entire period of spermatogenesis), yet others required a washout of just a few days. Key to resolving this fundamental difference in approach is achieving industry and regulatory consensus on whether information on spermatogenesis (eg, based on testicular pathology and testicular weights from repeat dose toxicity studies of at least 2-4 weeks’ duration) is sufficient to allow sponsors to shorten the duration of birth control to one linked to the duration of pharmacological activity rather than the duration of spermatogenesis. Banholzer et al 16 describe an option in the specific circumstance where a compound is genotoxic. This links the duration of birth control use to the duration of spermatogenesis, which is justifiable based on the need to avoid conception with sperm potentially damaged by a mutagen during mitosis or meiosis. However, in the absence of genotoxicity or overt effects on spermatogenesis, the scientific justification for requiring men to use birth control for months after the end of a trial is unclear. It is also worth noting that for many biologics that are not pharmacologically active in rodents and are using nonhuman primates for their nonclinical safety assessment, it is recognized that mating studies are generally not practical. 12
Duration of requirement for birth control following end of trials in WCBP
Most companies based duration of birth control for WCBP on the “five half-lives” described in the MHRA guidance. 2 However, as discussed in the commentary to part A, it was recognized that it would be preferred to base the duration of birth control use on more compound-specific consideration of the duration of time it takes for the clinical exposure level to drop below the exposure at the NOAEL for the embryo-fetal toxicity signal in all available studies, or below the NOEL for the pharmacological activity of concern. This concept is now reflected in the CTFG guidance document in the section titled “Definition of End of Relevant Systemic Exposure.”
Limited guidance on DDI data requirements
There appears to be limited industry or regulatory guidance for the DDI of concern for hormonal contraceptives. This topic would benefit from an evidence-based specialist review paper or consensus white paper.
Limited understanding of circumstances in which pregnancies arise in clinical trials
The survey indicated that no company conducts a systematic or geographic review of pregnancy rates across multiple clinical trials. The notes in ICH M3(R2) describe pregnancy rates from phase III studies to be <0.1% per menstrual cycle and even lower in phase II studies; this is quite different from the general WCBP population where unplanned pregnancies are common. 17
Based on current monitoring practices, many companies may not be able to spot downward or upward trends in pregnancy rates in clinical trials and would be unable to make evidence-based decisions on how to improve compliance and minimize pregnancy exposure in their clinical trials. It is also unclear if regulatory guidance (where it exists) on specific birth control methods for clinical trial settings has been written, taking adequate consideration of expert medical guidance and societal practices for that territory. For example, in the United Kingdom, there are specific National Institute for Health and Care Excellence guidelines on efficacy and use of long-acting reversible contraceptive (LARC) methods, 18 some of which have method failure rates approaching or lower than tubal ligation. 5 Many of these methods are much less prone to user error because they do not require daily or weekly compliance. However, despite these methods being log orders more efficacious and less prone to compliance problems, the MHRA guidance document 2 does not differentiate them from error-prone methods such as daily oral contraceptives.
Areas of Differences in Approaches to Clinical Trial Participation
One or 2 methods of contraception?
The survey revealed that some companies choose to adopt a single (strict) standard for the birth control requirements and management of WCBP in clinical trials whereas other companies choose to amend requirements depending on the existence of animal data and the results of those animal studies. For example, in the absence of embryo-fetal development data, or where there are signals of concern regarding developmental toxicity, some companies request the addition of a second birth control method to supplement the first method. Given the variability and lack of consistency in what is recommended as a primary method and the lack of interrogation of pregnancy rates across clinical trials, it is not possible to ascertain which approach is more effective at preventing pregnancies.
Role of patient choice
Where there is the intention of discouraging men from procreating when on a clinical trial, some companies implement that intention by instructing the men to wear a condom when having sexual intercourse. Other companies would instruct the participant not to father children (ie, either they or their partner should use contraception).
For trials with putative developmental toxicants, some companies permit the use of a single method of contraception and others do not. Where a woman has already chosen to use a LARC prior to enrollment in that clinical trial because she desires the high efficacy and convenience of that method, considering the inherent and real-world efficacy of LARC, 5,14,16 it is not clear how addition of a second method could improve the efficacy of her primary method of choice.
Some companies did not specifically state that they allow total sexual abstinence as an accepted method of birth control. The MHRA guidance document permits total sexual abstinence “only when it is the preferred and usual lifestyle choice” of the trial participant and the CTFG guidance requires that the reliability of sexual abstinence needs to be evaluated in relation to the duration of the clinical trial and the preferred and usual lifestyle of the subject. In those occasions where the health authority insists that sexual abstinence is only permitted where it is the usual lifestyle choice of the patient (despite sponsor submissions requesting a more flexible wording for a specific patient group), this in effect means that a woman who is incapacitated and/or sexually inactive through serious illness or surgery and has stopped taking contraceptives is ineligible for enrollment even in short-duration trials because sexual abstinence was not her preferred lifestyle choice in the weeks or months leading up to her serious illness or surgery. This anomaly may need resolving on a case-by-case basis.
Conclusions/Recommendations for the Future
This article identifies various practical issues that could be resolved through further scientific discussion between interested parties and relevant subject experts, ultimately leading to provision of company and regulatory guidance that is both pragmatic and patient centric. For many of the sampled respondents, there appears to be coordinated effort between nonclinical and safety functions; however, within some companies, these functions appear to operate independently, with little internal discussion and alignment on what the nonclinical reproductive toxicology data mean for the clinical risk assessment and how best to mitigate identified risks. Expert-led definitions regarding WCBP, birth control strategies for women in low-risk versus high-risk clinical trial situations, and practical DDI strategies would be particularly advantageous for industry. The scientific and evidence bases of the advice to male participants would also benefit from a fundamental debate about the nature of the underlying risks and how those are best managed both in clinical trials and post marketing. As well as providing a consistent framework for clinical trial management, such guidance may facilitate more consistent information about contraception in product labels, which, at the moment, is remarkably variable. 19 To facilitate these aims, scientists from US and European health authorities were invited to participate in the discussion of these survey responses.
During the scripting of this article, the CTFG of the HMA published advisory, nonbinding recommendations related to birth control and pregnancy testing in clinical trials. 4 This guidance provides a definition for WCBP and describes expectations regarding pregnancy testing and birth control regimes in different clinical trial situations. For high-risk situations, this guidance recommends use of a single highly effective method (from a list that concurs with those described by WHO 5 ) with a particular emphasis on those methods that have “low user dependency” such as hormone implants, IUDs, and other methods that are less prone to user error. The European guidance does provide recommendations beyond current industry practices by inferring the need for more clinical DDI testing, specifically with hormonal contraceptives, than might routinely be done. This thoughtful guidance provides clear recommendations but does not rule out case-by-case deviations from the core recommendations where the sponsor can provide specific justification. Further international discussion would be helpful in establishing global practices that are practical, patient centric, and robust.
Footnotes
Author Note
The opinions in this article are the personal views of the authors and do not reflect an official position of Amgen Inc, the Federal Agency for Medicines and Health Products, the Federal Institute for Drugs and Medical Devices, or of any European Medicines Agency committee or working party.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J Stewart, W Breslin, BK Beyer, K Chadwick, L De Schaepdrijver, M Desai, B Enright, JY Hui, G Moffat, and B Tornesi are employed by the pharmaceutical industry, from which the survey data presented in this manuscript were derived. There are no conflicts of interest for the remaining authors.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.