
Abstract
Significance:
Keloids represent a persistent clinical challenge, with recurrence rates approaching 100% after conventional surgical excision. Historically viewed as fibroblast-driven scars, emerging evidence positions immune dysregulation as a central driver of keloid pathogenesis, reshaping diagnostic and therapeutic paradigms.
Recent Advances:
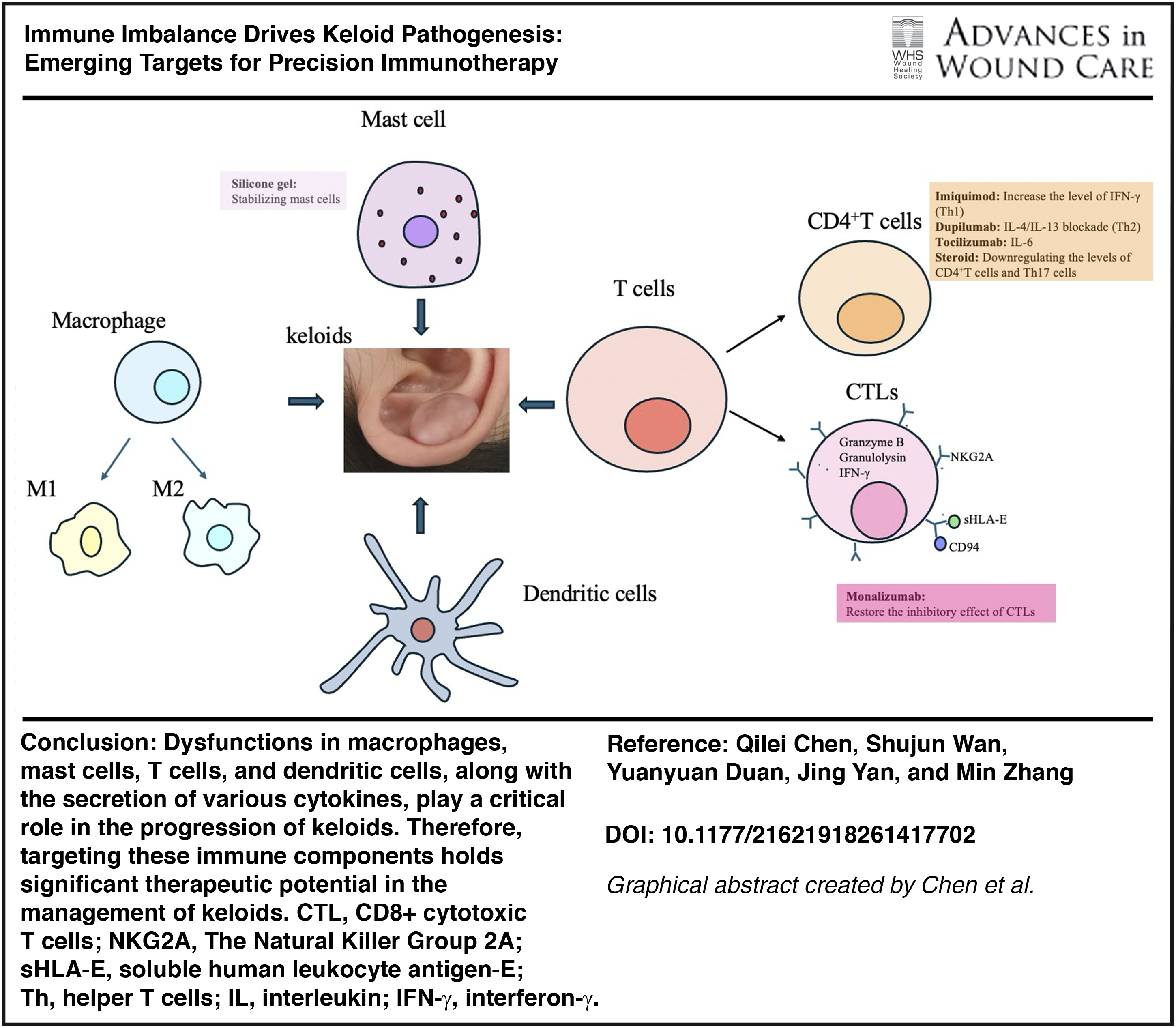
Multi-omics and single-cell analyses reveal profound alterations in immune cell populations and cytokine networks within keloid tissue. M2 macrophages, mast cells, Th2 and Th17 subsets, and dendritic cells dominate the inflammatory microenvironment, sustaining fibroblast activation and excessive extracellular matrix deposition. Key signaling axes—including transforming growth factor-β (TGF-β)/Smad, IL-4/IL-13, IL-6/JAK-STAT-3, and PI3K/AKT/mTOR—intersect with mechanical stress pathways, creating a self-perpetuating fibrotic loop. Notably, soluble human leukocyte antigen-E emerges as a predictive biomarker for disease progression and recurrence, while tissue-resident memory T cells may underlie postoperative relapse.
Therapeutic Implications:
Beyond corticosteroids and silicone gel, immune-targeted strategies are gaining traction. Dupilumab (IL-4/IL-13 blockade) demonstrates clinical efficacy in reducing keloid burden and pruritus. Experimental approaches targeting TGF-β signaling (Fresolizumab, AVID200), NKG2A/CD94 checkpoints (Monalizumab), IL-6 (Tocilizumab), and TSLP (Tezepelumab) hold promise for precision immunotherapy. Localized delivery and combination regimens may optimize outcomes while minimizing systemic toxicity.
Critical Issues and Future Directions:
The absence of validated keloid models and large-scale trials limits translation. Future research must integrate immune profiling, biomarker validation, and mechanistic modeling to enable personalized interventions. Immune dysregulation is not merely an epiphenomenon—it is the Achilles’ heel of keloids, offering unprecedented opportunities for targeted therapy and recurrence prevention.
Get full access to this article
View all access options for this article.