
Abstract
Background
This study evaluated the efficacy of montelukast in reducing seasonal allergic rhinitis symptoms in Japanese children with Japanese cedar (JC) pollinosis induced in an artificial exposure chamber (OHIO Chamber).
Methods
Pediatric patients aged 10 to 15 years sensitive to JC pollen entered a randomized, double-blind, single-site, crossover study. After confirmation of an allergic response to a JC pollen exposure for 3 hours in the OHIO Chamber during the screening period, subjects received either montelukast 5 mg chewable tablets or placebo for a 7-day treatment period, followed by a 3-hour pollen exposure in the chamber. After a 7-day washout period, subjects crossed over to the other treatment. Subjects were instructed to self-assess their nasal symptoms using 5-point scale for every 30 minutes. The primary end point was the change from baseline (just before entering the exposure chamber for each exposure) in total nasal symptom score (TNSS; the sum of nasal congestion, nasal discharge, and sneezing scores) over 3 hours of pollen exposure. Adverse events (AEs) were evaluated throughout the study.
Results
A total of 220 subjects (median age, 12 years) received treatment. For TNSS, the between-group difference in the change (95% confidence interval) was −0.01 (−0.11 to 0.10); the change between placebo and montelukast 5 mg was not significant. TNSS in the screening and treatment periods after receiving placebo for 7 days was 1.58 and 1.31, respectively, suggesting a placebo response. On account of high placebo response, a post hoc analysis was conducted. The analysis in a subgroup of subjects who did not show placebo response demonstrated a difference in the efficacy between montelukast and placebo (nominal P < .037). The most common AE was positive urine protein (4.6% with montelukast vs 7.8% with placebo).
Conclusions
Although montelukast was well tolerated, this study did not demonstrate a treatment difference between active drug and placebo in Japanese children exposed to JC pollen in the OHIO Chamber.
Keywords
Introduction
Allergic rhinitis (AR) is a disorder with symptoms such as sneezing, rhinorrhea, and nasal congestion and causes sleep disturbances, which can have a profound effect on the quality of life for pediatric patients as well as for their parents.1–4 Japanese cedar (JC) pollinosis has contributed to a large increase in the prevalence of AR in Japan and induces particularly severe symptoms of AR. 5
The pharmacologic targeting of the inflammatory mediators, cysteinyl leukotrienes, has demonstrated efficacy in the treatment of AR.6–8 Cysteinyl leukotrienes are generated from mast cells, eosinophils, and basophils during allergic reactions. The clinical manifestations of the effect of cysteinyl leukotrienes include AR symptoms such as nasal obstruction, nasal discharge, and sneezing.9,10 Montelukast is a selective antagonist of cysteinyl leukotriene receptor type 1 with demonstrated efficacy in AR. 8
We conducted a crossover trial to evaluate the efficacy, safety, and tolerability of montelukast, administered as 5 mg chewable tablets (CT), in Japanese children with JC pollinosis. Evaluating pharmacologic treatments for AR can be difficult due to environmental differences among various locations and weather pattern variations from year-to-year, which affect pollen counts and/or exposure. We used an allergen exposure chamber (OHIO Chamber) in this study to allow for exposure to a fixed number of pollen grains in a stable setting, irrespective of variable environmental factors. 11
Methods
This study (Clinical Trials Registry # NCT01852812, Sponsor Protocol MK-0476-519) was conducted at 1 trial center in Japan from June 2013 to September 2013, which is outside of the pollen season. The study protocol was approved by an institutional review board of the Shinanozaka Clinic (Tokyo, Japan) and was conducted according to principles of Good Clinical Practice. The parent or legal guardian provided written informed consent for the subject’s participation in the trial; subject assent was additionally obtained whenever possible.
Subjects
Japanese male and female children between the ages of 10 and 15 years (inclusive), ≥25 kg in weight, and ≥125 cm in height were enrolled. Female subjects who had begun menstruating were required to have a negative beta-human chorionic gonadotropin pregnancy test and agreed to remain abstinent from heterosexual activity or use 2 adequate barrier methods of contraception to prevent pregnancy during the trial.
Subjects were sensitive to only JC pollen (positive JC pollen-specific IgE-antibody assay [allergen score ≥2] or a positive skin test for JC pollen at screening visit) and had AR symptoms during at least the previous 2 pollen seasons with dominant symptom of nasal congestion. AR caused by any other allergen except JC pollen led to exclusion from the study. Specific exclusion criteria are in Supplement 1.
Study Design
This was a randomized, placebo-controlled, crossover, single-center, double-blind trial of montelukast 5 mg CT conducted in nonpollen season. The study design is illustrated in Figure 1. To demonstrate baseline sensitivity to JC pollen, all subjects underwent a screening JC pollen exposure at visit 2 after receiving single-blind placebo for 7 days. They were exposed to a fixed concentration (8000 grains/m3) of JC pollen for 3 hours under a controlled temperature and humidity in the OHIO Chamber as described elsewhere. 12 They were instructed to rate their symptoms every 30 minutes according to severity scoring criteria (0–4 point scale). Only the subjects who increased by at least 1 point on the nasal congestion, nasal discharge, or sneezing score on 1 occasion (30, 60, 90, 120, 150, and 180 min) during the allergen exposure relative to each recorded score before entering the chamber were eligible to proceed to the treatment phase. Randomization of treatment sequence occurred at visit 3 according to a computer-generated randomization schedule; subjects were assigned in a 1:1 ratio to a sequence in which they received montelukast 5 mg CT once daily as the first period followed by matching placebo for 7 days with a washout period of 7 days or to a sequence in which they received matching placebo first followed by montelukast 5 mg CT once daily in the same manner. The treatment period was performed in a double-blind fashion. On the last day of each treatment period, subjects were exposed to JC pollen at a concentration of 8000 grains/m3 for 3 hours and rated their nasal symptoms for every 30 minutes during pollen exposure. Following visit 6, the investigator or trial staff contacted the subjects’ caregivers by phone for up to 14 days after the last dose of study medication to determine whether any serious adverse events (AEs) had occurred since the posttrial clinic visit (Figure 1). Details of the timing of all procedures are provided in Supplement 2.

Study design.
Efficacy Measurements
Efficacy was evaluated by assessing nasal symptom scores that were rated by subjects according to severity based on a 5-grade classification system (score 0–4) during each 30-minutes interval (Supplementary Table 1). The primary end point was the change from baseline in the total nasal symptom score (TNSS), defined as the sum of 3 scores measuring nasal congestion, nasal discharge, and sneezing, averaged across all timepoints during 3 hours. Secondary end points included the change from baseline in the weighted TNSS (weighted 2:1:1 for nasal congestion, nasal discharge, and sneezing, respectively) averaged across all timepoints during 3 hours (the weighted approach was prespecified for analysis due to significant effects on nasal congestion observed with leukotriene antagonists), the change from baseline in each individual nasal symptom score averaged across all timepoints during 3 hours, the change from baseline in the TNSS at each individual timepoint (30, 60, 90, 120, 150, and 180 min), the change from baseline in the weighted TNSS at each timepoint, and the change from baseline in each nasal symptom score at each timepoint.
Patient Characteristics.

Abbreviations: SAR, seasonal allergic rhinitis; SD, standard deviation.
A post hoc, exploratory analysis of the changes from baseline in TNSS averaged during 3 hours of exposure in subject groups by age range (10- and 11-year-old group, 12- and 13-year-old group, and 14- and 15-year-old group) was performed. An additional subgroup analysis was performed to evaluate changes from baseline in TNSS in subjects who did not exhibit a placebo response (ie, a clinically meaningful change in TNSS between the screening placebo period and the placebo sequence during the double-blind treatment period). A clinically meaningful change was defined as a change of 1 point out of a total of 6 points in the TNSS score; this was calculated as a change of 0.17 (ie, 1/6) in the TNSS score between the screening placebo period and the treatment placebo sequence.
The number of nasal discharge eosinophils was also evaluated in those patients who had nasal discharge at the end of pollen exposure. Nasal secretions were collected from the subject after the allergen exposure. The number of nasal discharge eosinophils was counted by the central laboratory (SRL, Inc., Tokyo, Japan) and categorized as—(none detected), 1+ (a few ∼ detected as scattered/all high-power field [HPF]), 2+ (a few/each HPF), and 3+ (detected as clustered) under a microscopic finding.
Safety Evaluation
AEs were monitored throughout the study and were evaluated for severity and relationship to study medication by the investigator. Safety and tolerability were assessed by measuring the percentage of subjects with clinical AEs or changes in laboratory values.
Statistical Analysis
The full analysis set (FAS) population served as the primary population for the analysis of efficacy data. The FAS population consisted of all randomized patients who took at least 1 dose of study treatment and had at least 1 postbaseline observation. The primary hypothesis was evaluated using a longitudinal data analysis model (analysis of covariance mixed effects model with TNSS at baseline [before entering the chamber room in each period] as a covariate, sequence, treatment, and period as fixed effects and subject as a random effect) on the change from baseline of TNSS. The mean change from baseline in the TNSS was estimated, and the treatment difference between montelukast and placebo was evaluated with 95% confidence intervals (CI) and P values.
An all patients as treated (APaT) approach was used for the safety analyses. The APaT population consisted of all randomized patients who received at least 1 dose of study treatment. Safety and tolerability were assessed by a clinical review of all relevant parameters including AEs and laboratory safety parameters. AEs, drug-related AEs, serious AEs, serious drug-related AEs, discontinuation due to AEs, and AEs occurring in 4 or more subjects in any treatment group were assessed via point estimates with 95% CIs for between-treatment comparisons. The 95% CI was calculated using the asymptotic method. Point estimates by treatment group were provided only for the AEs that developed in fewer than 4 subjects in each treatment group and changes from baseline in laboratory parameters.
This study was planned to randomize 220 subjects (110 subjects per sequence). It had 90% power for 220 subjects to demonstrate that montelukast was superior to placebo at a 2-sided 5% alpha level. The power and sample size calculations were based on the following assumptions: an underlying treatment difference of 0.23, a within-subject standard deviation (SD) of 0.7, and dropout rate of approximately 10%.
Results
Subjects
A total of 220 subjects were enrolled in this study; 110 subjects were randomized to sequence 1 (montelukast/placebo) and the other 110 to sequence 2 (placebo/montelukast). Patient demographics for each sequence were similar and are shown in Table 1. Of the 220 subjects, 208 completed the study, and the reasons for withdrawn are shown in Figure 2.

Disposition of subjects.
Efficacy
The change from baseline in the TNSS when averaged during 3 hours of exposure with montelukast treatment (n = 213) did not differ from that with placebo treatment (n = 211) (primary end point). The change from baseline in the TNSS when averaged during 3 hours of exposure with montelukast (n = 213) and with placebo treatment (n = 211) was 1.17 and 1.18, respectively. The between-treatment difference for the change from baseline (95% confidential interval) was −0.01 (−0.11 to 0.10), which was not significant (Table 3 and Figure 3).
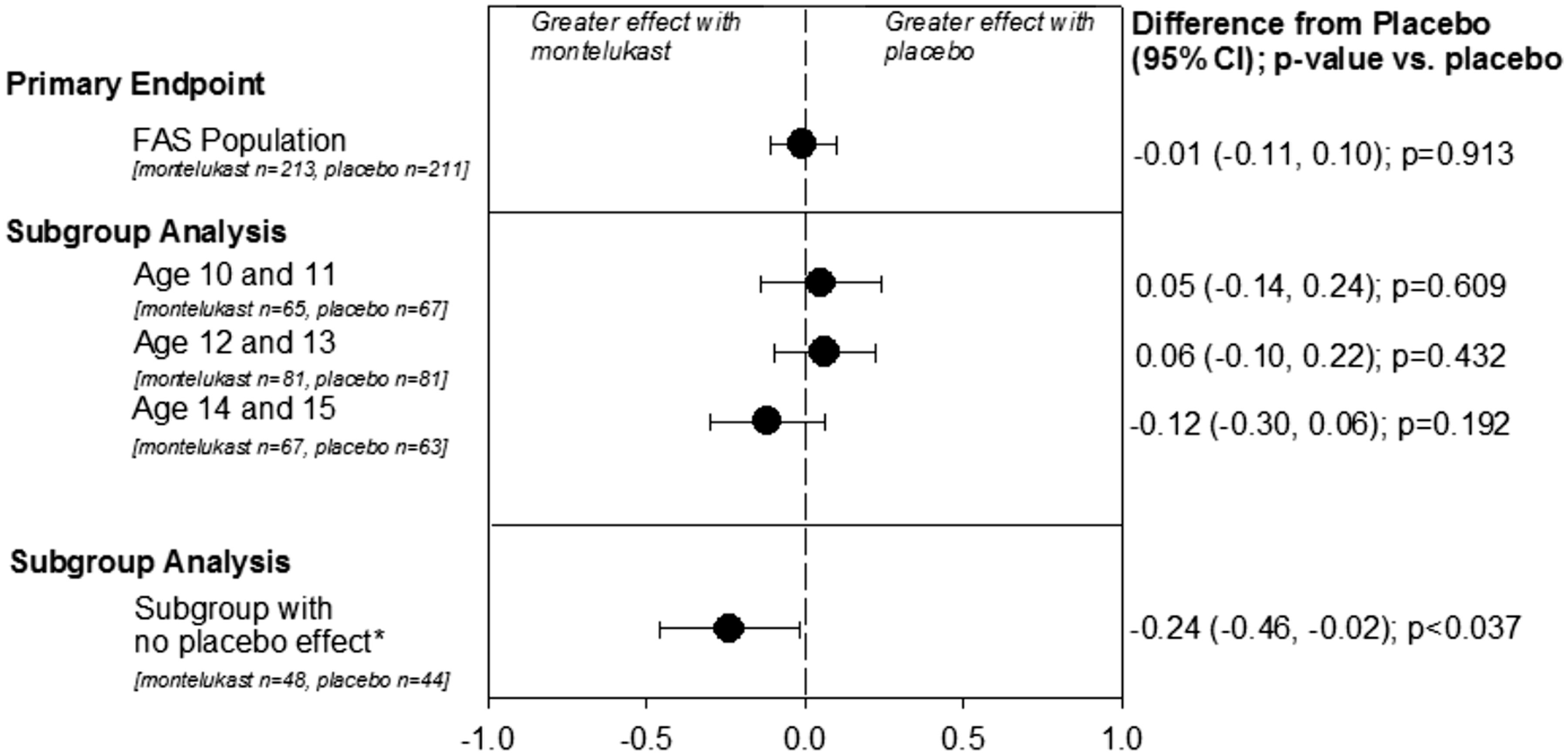
Efficacy summary: change from placebo for primary end point (mean TNSS over 3 h) and subgroup analyses by age and for patients who did not exhibit a placebo effect (FAS population). Based on longitudinal data analysis model with baseline TNSS as a covariate, sequence, treatment, and period as fixed effects and subject as a random effect. The values in P for post hoc subgroup analyses were nominal because they were not adjusted for multiplicity. FAS, full analysis set. *No clinically important difference (ie, change in 1 point on 6-point TNSS scale) between the screening placebo run-in period and the placebo sequence of the treatment period.
The secondary end point of change from baseline of the weighted TNSS (total of nasal congestion, nasal discharge, and sneezing scores weighted in the ratio of 2:1:1) averaged during 3 hours of exposure with montelukast treatment did not differ from that with placebo treatment. The between-treatment difference for the change from baseline (95% CI) was −0.00 (−0.17 to 0.16), which was not significant (Table 3).
The between-treatment differences (95% CI) in changes from baseline for each individual nasal symptom score averaged during 3 hours of exposure with montelukast treatment were −0.00 (−0.07 to 0.07) for nasal congestion, −0.03 (−0.06 to 0.01) for nasal discharge, and 0.02 (−0.01 to 0.05) for sneezing. The variation in the changes from baseline of the TNSS, weighted TNSS, and each individual nasal symptom score at 30, 60, 90, 120, 150, and 180 minutes after entering the chamber revealed that the changes with montelukast were not significantly lower than those with placebo at any time point (data not shown).
Samples of nasal discharge eosinophils were taken only from subjects with nasal discharge at the end of the exposure; therefore, samples were not available for 50.2% (108/215 subjects) of the montelukast treatment group and 45.1% (97/215 subjects) of the placebo treatment group.
In subjects in the category of patients with the highest level of detectable eosinophils (3+), fewer subjects had nasal discharge eosinophils with montelukast treatment (15.9%, 17 of the 107 subjects) compared with placebo treatment (27.1%, 32 of the 118 subjects) (Table 2).
Levels of Nasal Discharge Eosinophilic After 3-hour Exposure.

aProportion of subject with nasal discharge assessed.
Summary of Efficacy (Change From Baseline for the Primary End Point and Subgroup Analyses).

Abbreviations: CI, confidence interval; LS, least square; SD, standard deviation; TNSS, total nasal symptom score.
Nasal congestion, nasal discharge, and sneezing scores weighted in the ratio of 2:1:1.
No clinically important difference (ie, change in 1 point on 6-point TNSS scale) between the screening placebo run-in period and the placebo sequence of the treatment period.
The mean (SD) change from baseline in TNSS during the screening placebo run-in period (n = 216) was 1.58 (1.09) (mean at baseline: 0.12; 3 h: 1.70). The mean (SD) TNSS during the treatment period after 7 days of montelukast (n = 213) was reduced to 1.29 (1.05). However, mean (SD) TNSS was 1.32 (1.10) after patients received placebo during the treatment period (n = 211), indicating a placebo effect. Of note, when observing each period regardless of treatment, the mean (SD) TNSS was 1.68 (1.03), 1.32 (1.04), and 1.25 (1.05) for the screening placebo run-in period, period 1, and period 2, respectively.
For the subgroup analysis of subjects by age, the differences in TNSS between the montelukast group and the placebo group (95% CI) were 0.05 (−0.14 to 0.24), 0.06 (−0.10 to 0.22), and −0.12 (−0.30 to 0.06) in the 10- to 11-year-old groups (n = 65 and 67 in montelukast and placebo, respectively), 12- to 13-year-old groups (n = 81 and 81), and 14- to 15-year-old groups (n = 67 and 63), respectively (Table 3 and Figure 3).
In the post hoc analysis of the subgroup of subjects who had no clinically meaningful change in TNSS between the screening placebo run-in period and the placebo sequence of the treatment period (n = 48 and 44 in montelukast and placebo, respectively), there was a between-group difference of −0.24 (−0.46 to −0.02) (nominal P < .037) that demonstrated a treatment effect for montelukast versus placebo (Table 3 and Figure 3).
Safety and tolerability
AEs were observed for 10.2% of the montelukast group and 16.5% of the placebo group (Table 4). The most common AEs were positive urine protein, abnormal hepatic function, and increased alanine transaminase (ALT). These AEs were mild in intensity and did not result in treatment discontinuation. There were no serious AEs in this study, and only 1 subject in the montelukast group discontinued due to an AE (moderate, nondrug-related nasopharyngitis). There were no clinically meaningful variations in the change from baseline of the laboratory test results. Of note, although a numerical difference in proteinuria was observed with 4.6% (10/216 points) of montelukast patients and 7.8% (17/218 points) of placebo patients who were positive for urine protein, this difference was not statistically significant according to a post hoc analysis (significance level α = 0.05).
Summary of AEs—Subjects With AEs (APaT).
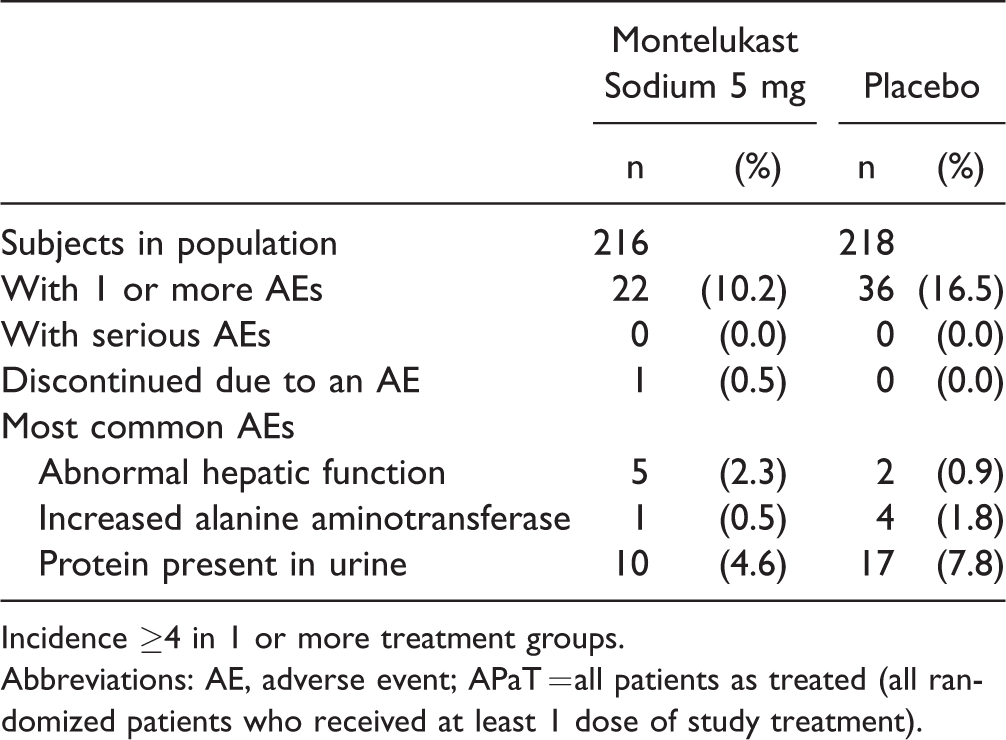
Incidence ≥4 in 1 or more treatment groups.
Abbreviations: AE, adverse event; APaT = all patients as treated (all randomized patients who received at least 1 dose of study treatment).
Discussion
The results of this trial showed no significant differences between montelukast and placebo for the treatment of JC pollinosis induced by an artificial exposure chamber in Japanese children aged 10 to 15 years. The mean TNSS over 3 hours changed from 1.70 during the placebo run-in period to 1.29 during the montelukast treatment period; however, the mean TNSS over 3 hours during the placebo treatment period changed to a similar degree to 1.32 demonstrating a strong placebo response. This strong placebo response during the treatment period hindered the ability to measure the effectiveness of montelukast in this population of children with JC pollinosis.
Previous studies have demonstrated the efficacy of montelukast in AR in adult and older adolescent populations (aged 15–65 years) exposed repeatedly to daily natural airborne pollen during at least 2 weeks of treatment during a pollen season.13–18 Rather than natural exposure, the present study used just 3 hours of exposure in an exposure chamber, a validated method to evaluate AR that was developed to reduce seasonal and environmental variability. 11 A previous, similarly designed chamber study in JC pollinosis demonstrated a significant difference in TNSS for the leukotriene receptor antagonist pranlukast versus placebo although the change from baseline in TNSS with pranlukast was similar to that observed with montelukast in our study. 19 Conflicting data have demonstrated a lack of treatment efficacy with pranlukast in children with AR in a field study (ONO-1078-36) and a chamber study (ONO-1078-40). 20 These results highlight the difficulty of evaluating treatment for AR in a pediatric population.
A single-blind run-in was implemented and we used a crossover design against a parallel-group design to reduce variability, although a limitation of crossover designs is that the duration of treatment is shortened. In this study, the treatment duration was 7 days, which may not have been sufficient to observe the efficacy previously shown with 2 weeks of montelukast treatment during JC season. Further, this study evaluated AR symptoms in a controlled laboratory setting in which efficacy is evaluated for only up to 180 minutes after pollen exposure rather than in a real-world setting in which inflammation increases with repeated daily exposure to pollen over weeks during a season. Therefore, this study may not adequately replicate the effects of montelukast in a natural environment.
Measures were taken to reduce a possible placebo effect in this study. But the use of subjective end points in pediatric subjects may have been a limitation that contributed to a placebo effect. To explore the effect of younger age in the ability to provide reliable subjective evaluations, we conducted subgroup analyses by age. Although no significant treatment-group difference was observed in any age subgroup, we noted a trend of greater treatment response in older subjects compared with the younger subjects. We also conducted a subgroup analysis to observe TNSS in patients who did not exhibit an apparent placebo effect (ie, a clinically meaningful difference between their screening placebo results and their treatment period placebo sequence results). An improvement was observed with montelukast versus placebo in this subgroup. These results are from an exploratory post hoc analysis conducted to better characterize the placebo response in this study and should be viewed with caution. Whereas the statistical interpretation of these post hoc analyses are limited due to no adjustment being done for multiplicity, these analyses may inform future study and suggest that a treatment effect can be observed if the placebo effect can be controlled.
The use of objective measures of AR would be an important advancement in the study of this condition, particularly in pediatric patients. The detection of eosinophils in nasal smears is an important diagnostic criterion for AR in Japan. 21 The number of nasal discharge eosinophils as a biomarker for eosinophil infiltration and inflammation has previously been demonstrated in pediatric patients with JC pollinosis.22,23 In the present study, we evaluated nasal discharge eosinophils in patients who had nasal discharge following pollen exposure (this included roughly half of all randomized patients), and a notable improvement with montelukast versus placebo was observed in this subgroup.
The evaluation of safety and tolerability demonstrated that patients receiving montelukast had no clinically meaningful differences in AEs or changes in vital signs and laboratory values compared with patients receiving placebo; there were numerically lower proportions of patients in the montelukast group with AEs compared with patients in the placebo group. There were no serious AEs observed in this study.
Conclusion
This study did not demonstrate a treatment difference between montelukast and placebo in pediatric patients aged 10 to 15 years with JC pollinosis after exposure to JC pollen in an artificial exposure chamber. This result may be due to a number of methodological causes such as high placebo response, the challenges to assess AR in young age patients, and the short duration of treatment. The analysis in a subgroup of subjects who did not show placebo response demonstrated a significant difference in the efficacy between montelukast and placebo (P < .037). These findings illustrate the challenges associated with the use of subjective end points in pediatric populations and highlight the need for age-appropriate measures to accurately assess AR symptoms in children. The more objective exploratory end point using the nasal discharge eosinophil suggested favorable improvement after montelukast treatment. Notably, the results of this study demonstrated that the chamber study design in this pediatric population was safe and well tolerated.
Supplemental Material
Supplemental material for Evaluation of Montelukast for the Treatment of Children With Japanese Cedar Pollinosis Using an Artificial Exposure Chamber (OHIO Chamber)
Supplemental material for Evaluation of Montelukast for the Treatment of Children With Japanese Cedar Pollinosis Using an Artificial Exposure Chamber (OHIO Chamber) by Kazuhiro Hashiguchi, Kimihiro Okubo, Yoichi Inoue, Hirotaka Numaguchi, Kumi Tanaka, Nobuyuki Oshima, Anish Mehta, Chisato Nishida, Itori Saito, George Philip in Allergy & Rhinology
Footnotes
Acknowledgments
This study was designed by MSD K.K., Tokyo, Japan, a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Statistical analyses were performed by the Clinical Biostatistics department of MSD K.K. and Merck & Co., Inc., Kenilworth, NJ, USA. The authors thank Christine McCrary Sisk and Jennifer Pawlowski (both from Merck & Co., Inc., Kenilworth, NJ, USA) for editorial and administrative assistance.
Ethical Approval
This study was approved by our institutional review board (Shinanozaka Clinic, Tokyo, Japan).
Statement of Human and Animal Rights
This study was conducted according to principles of Good Clinical Practice.
Statement of Informed Consent
The parent or legal guardian provided written informed consent for the subject's participation in the trial; subject assent was additionally obtained whenever possible.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: K.H. reports no conflicts of interests. K.O. has received research support from Torii Pharmaceutical Co., Ltd as well as consulting and/or lecture fees from the following: Torii Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., MSD K.K., Teikoku Seiyaku Co., Ltd., Hisamitsu Pharmaceutical Co., Inc., Ono Pharmaceutical Co., Ltd., Taiho Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, GlaxoSmithKline K.K., Daiichi Sankyo Co., Ltd, Kyorin Pharmaceutical Co., Ltd., Eisai Co., Ltd., Sumitomo Dainippon Co., Ltd., Nichi-Iko Pharmaceutical Co., Ltd., Nippon Shinyaku Co., Ltd., Sanofi K.K. Y.I., H.N., K.T., N.O., C.N., and I.S. are employees of MSD K.K. and may own stock or stock options in the company. A.M. and G.P. are employees of Merck & Co., Inc., Kenilworth, NJ, USA and may own stock or stock options in the company.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Merck & Co., Inc., Kenilworth, NJ, USA.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.