
Abstract
Sinus of Valsalva aneurysm (SVA) is an abnormal dilatation of the aortic root located between the aortic valve annulus and the sinotubular junction and is rare in the pediatric population. This case report describes a unique case of a 16-year-old adolescent patient admitted with progressive heart failure symptoms and diagnosed with a ruptured noncoronary SVA. He underwent surgical repair of the SVA with autologous pericardial patches and had an uncomplicated postoperative course. A genetic workup revealed an underlying 22q11.2 deletion that is infrequently associated with SVA.
Introduction
Sinus of Valsalva aneurysm (SVA) is caused by weakness of the elastic tissue between the aortic valve annulus and the sinotubular junction resulting in an aneurysmal dilatation.1,2 Sinus of Valsalva aneurysms can be congenital due to connective tissue disorders like Marfan or Ehlers-Danlos syndrome or acquired due to bacterial endocarditis, syphilis, tuberculosis, vasculitis, trauma, or atherosclerosis.2-4 Sinus of Valsalva aneurysm is rare in the pediatric population and accounts for only 0.1% to 3.5% of congenital heart disease cases. 5 We report a previously healthy 16-year-old boy with a ruptured noncoronary SVA into the right atrium. He was diagnosed postoperatively with 22q11.2 deletion syndrome (22q11.2DS) that is not commonly associated with SVA.
Case Report
A 16-year-old 85 kg boy with no significant past medical history was admitted to the pediatric cardiac intensive care unit with progressively worsening heart failure symptoms of fatigue, bilateral leg swelling, and orthopnea for one month. On presentation to the emergency room, he was hypertensive to 160/80 mm Hg and tachycardic to 100 beats/min with a grade III holosystolic murmur best appreciated in the left sternal border and normal oxygen saturations on room air. The remainder of his physical examination was significant for hepatomegaly and bilateral pitting pedal edema. A chest x-ray showed cardiomegaly with evidence of interstitial pulmonary edema, and an electrocardiogram showed ventricular bigeminy and inverted T waves in V5, V6. Pro-brain natriuretic peptide was elevated to 1880 pg/mL but troponin was normal at 10 ng/L. Inflammatory markers were normal. A transthoracic echocardiogram (TTE) showed aortic noncoronary SVA rupture into the right atrium measuring 2 cm in diameter, with left to right shunting (Figure 1), mild aortic valve regurgitation and aortic root dilatation with a diameter of 4 cm (z-score 3.01). A cardiac computed tomography confirmed the TTE findings with no evidence of aortic arch or coronary anomalies (Figure 2A-D). He underwent surgical repair of the SVA through a median sternotomy. Intraoperatively, upon right atriotomy, a very large fenestrated aneurysmal tissue was identified just above the anterior septal commissure of the tricuspid valve, that was excised, and the defect was repaired with two layers of autologous pericardial patches (Figure 3A and B). An intraoperative transesophageal echocardiogram revealed preserved biventricular function with trivial aortic regurgitation. He was extubated on postoperative day 1 and after an uncomplicated postoperative course, he was discharged home on postoperative day 8 on lisinopril and furosemide. He is doing well on a two-month and a six-month outpatient cardiology follow-up with no recurrence of heart failure symptoms and has been cleared to take part in competitive sports. A rapid whole exome sequencing analysis sent to rule out an underlying genetic etiology, revealed an 846 kb deletion at 22q11.2 consistent with a diagnosis of 22q11.2DS. Of note, he had no evidence of dysmorphic features on examination with no early developmental delays or learning disabilities.
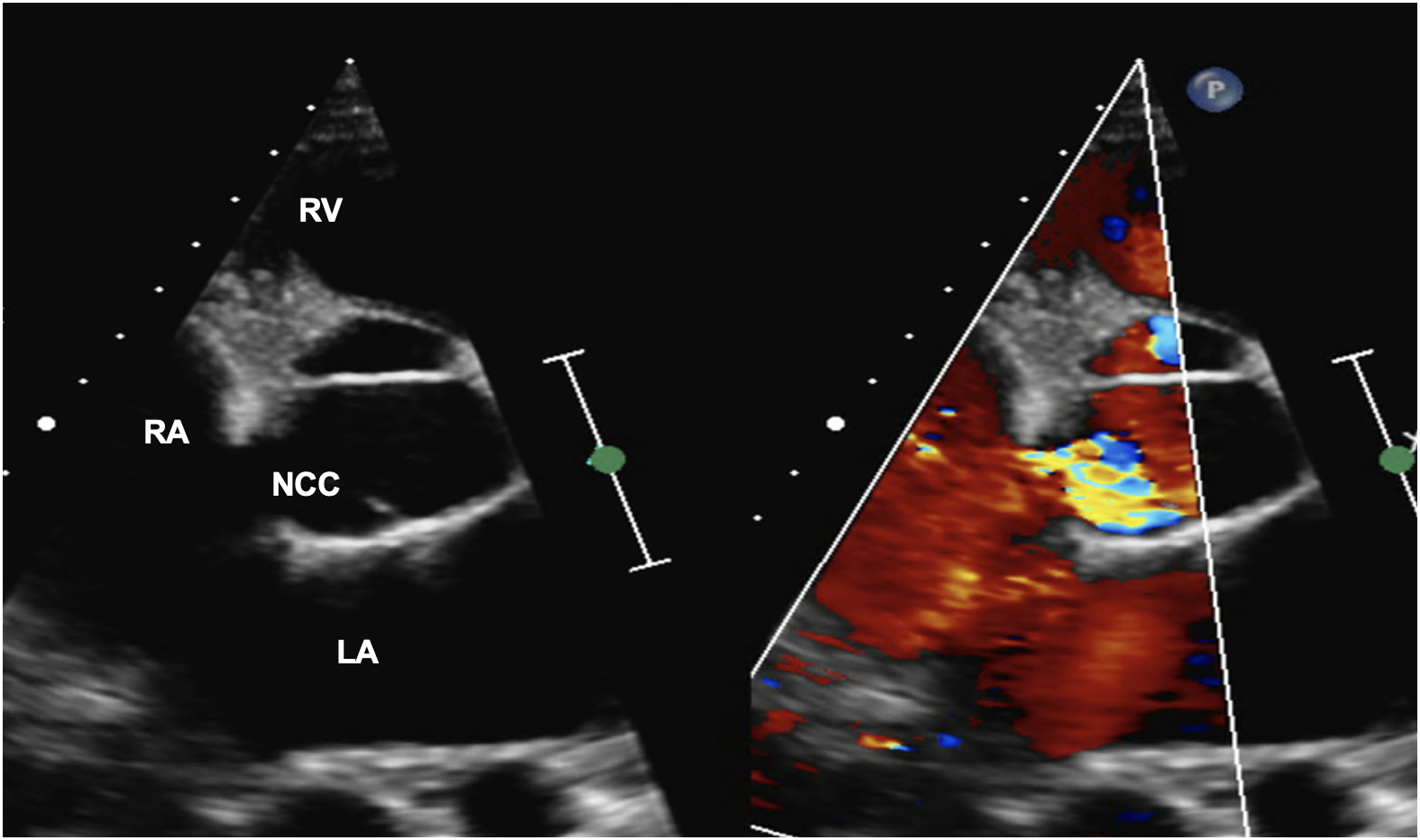
TTE parasternal short axis view with color Doppler showing the communication from the noncoronary aortic sinus to the right atria with a significant left to right shunt. LA, left atrium; NCC, noncoronary cusp; RA, right atrium; RV, right ventricle.
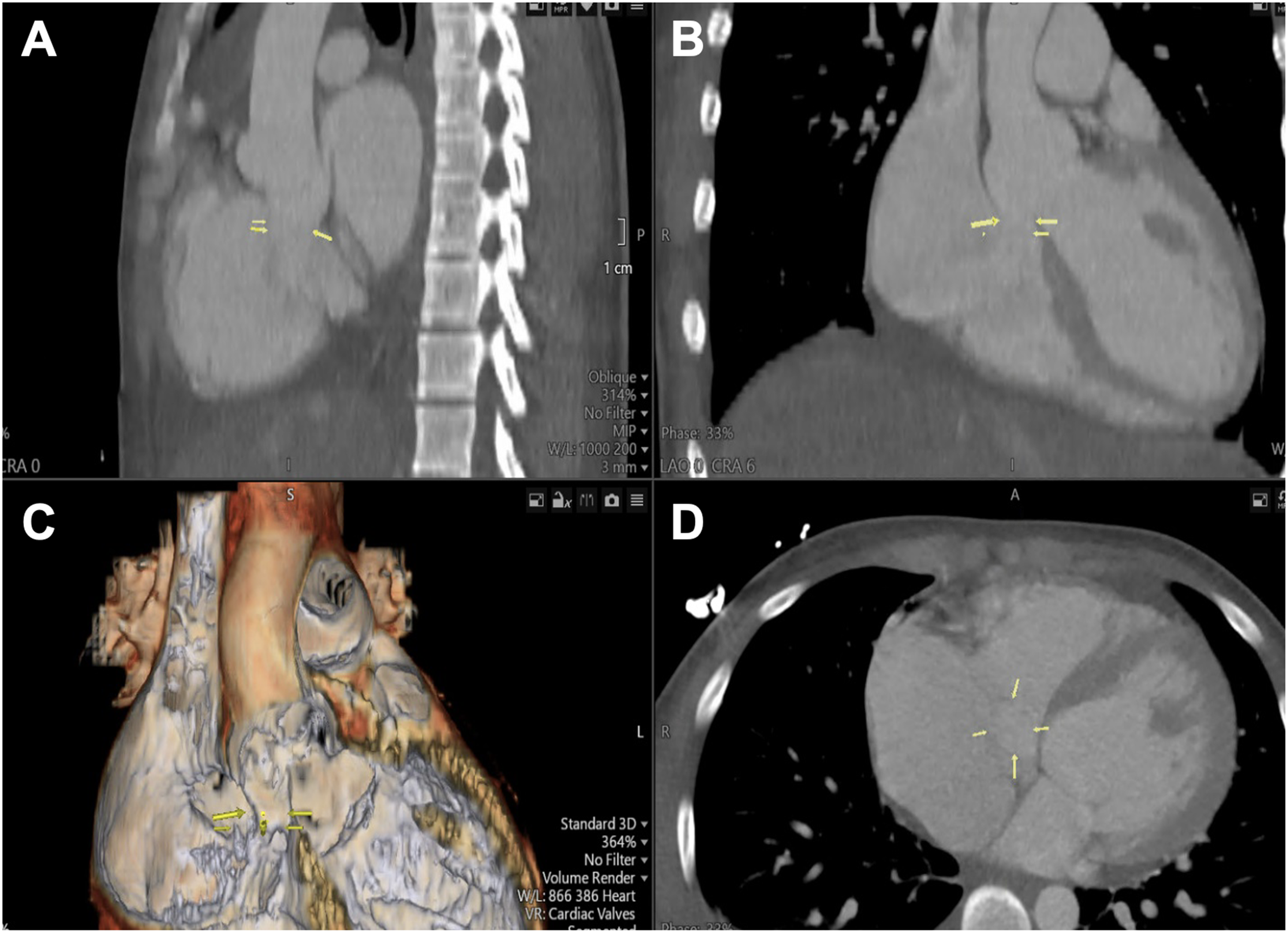
Computed tomography (CT) angiogram (A-D) showing the communication between the aortic root and the right atrium measuring 2 cm (yellow arrow).
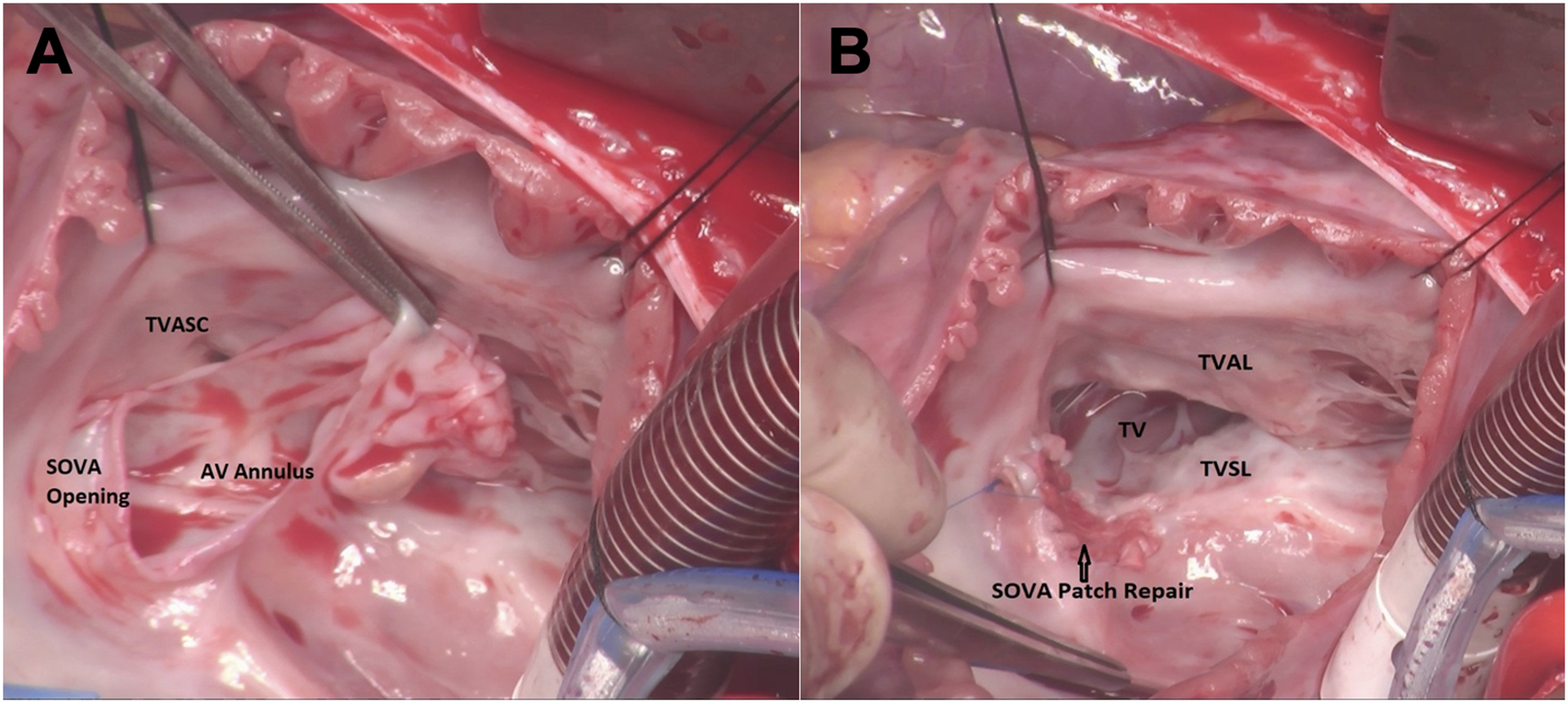
Intraoperative view of a large fenestrated aneurysmal tissue (A) that was repaired with two layers of autologous pericardial patches (B). AV, aortic valve; SOVA, sinus of Valsalva aneurysm; TV, tricuspid valve; TVAL, tricuspid valve anterior leaflet; TVASC, tricuspid valve anteroseptal commissure; TVSL, tricuspid valve septal leaflet.
Discussion
The clinical presentation of SVAs is dependent on its size, location, and the chamber into which it ruptures. They originate mostly from the right coronary sinus (75%), followed by the noncoronary sinus (20%), and rarely from the left coronary sinus (<5%). The right ventricle followed by the right atrium is the most common locations of rupture.3,4 Intact SVAs are mostly asymptomatic but can rarely cause aortic regurgitation, cardiac arrhythmias, and acute coronary syndrome from thrombotic occlusion of the coronary ostia.1,5 Sinus of Valsalva aneurysm rupture typically occurs between 20 and 40 years of age and has been reported to occur in less than 15% of pediatric patients with SVA. If left unrecognized, it can cause serious complications such as progressive congestive heart failure, refractory cardiogenic shock, pericardial tamponade, and death.1,3 Our patient's clinical course emphasizes the significance of early detection of an SVA, as patients tend to have excellent outcomes when they undergo intervention early. 1 Surgical repair is indicated for ruptured aneurysms or an unruptured symptomatic aneurysm. Percutaneous transcatheter closure has also been reported as a surgical alternative. There are no randomized controlled trials comparing the two approaches. 1 A recent meta-analysis of 330 patients from 10 studies showed that percutaneous transcatheter closure was associated with a shorter length of hospital stay but had no statistically significant differences in in-hospital mortality, incidence rates of postinterventional residual shunts, or aortic regurgitation. 6 A surgical approach for SVA repair in our patient who had worsening heart failure symptoms and a high ectopy burden proved to be the best option because it resulted in a good postoperative outcome.
22q11.2 deletion syndrome is caused by a microdeletion on the long arm of chromosome 22. Cardiovascular defects are present in 80% of the patients with 22q11.2DS in infancy and is often the initial presentation. Tetralogy of Fallot, double-outlet right ventricle, truncus arteriosus, interrupted aortic arch, and malaligned ventricular septal defects are more common defects while progressive aortic root dilations have also been infrequently linked to 22q11.2DS. 7 To our knowledge, there have been only two previous case reports of SVA rupture associated with 22q11.2DS. 8 Unlike our index patient who was previously healthy, both patients who presented with SVA rupture had a classic DiGeorge syndrome phenotype. With reports of more uncommon etiologies of SVA, 9 a comprehensive genetic workup is warranted, even in patients with a normal phenotype, as they help stratify the risk of further cardiac and noncardiac manifestations of the underlying genetic syndrome.
Conclusion
Sinus of Valsalva aneurysm rupture is rare in children but has an excellent surgical outcome if recognized and intervention is performed early. In addition to more common conotruncal anomalies, 22q11.2DS has also been linked to progressive aortic dilatation, and SVA rupture as illustrated in our index patient.
Footnotes
Authors’ Note
Drs Aravinth P. Jawahar and Bayan Issa contributed equally to the article. All authors contributed to the case report by: (1) conception and design, acquisition of data or analysis and interpretation of data; (2) drafting the article or revising it critically for important intellectual content; (3) final approval of the version published; and (4) agreement to be accountable for the article and to ensure that all questions regarding the accuracy or integrity of the article are investigated and resolved.
Authors’ Statement
The authors affirm that consent to publish this report was obtained from the patient's parent.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.