
Abstract
Acquired angioedema due to C1-inhibitor deficiency (AAE-C1INH) is a rare condition characterized by the localized swelling of the deeper skin layers and mucous membranes, especially the face, lips, tongue, throat, and gastrointestinal tract. AAE-C1INH is strongly associated with lymphoproliferative disorders, although it can also be linked to autoimmune conditions, solid tumors, infections, or even occur without an identifiable cause. We present the case of a 45-year-old female patient with complaints of recurrent abdominal pain, bloating, and joint swelling. Laboratory testing showed decreased C1q and C4 complement levels, and C1 esterase inhibitor levels, indicative of AAE-C1INH. Further work up confirmed a diagnosis of extranodal marginal zone lymphoma with involvement of the bone marrow and spleen. Treatment with rituximab led to resolution of angioedema symptoms and almost complete remission of underlying lymphoma. This case underscores the importance of evaluating an underlying lymphoproliferative disorder in AAE-C1INH. Therefore, the early participation of a multidisciplinary team including specialists in immunology, hematology, and oncology is necessary for appropriate management.
Keywords
Introduction
Acquired angioedema due to C1-inhibitor deficiency (AAE-C1INH) is a rare condition in which the skin, gastrointestinal tract, and upper airway develop recurrent, non-itchy swelling. It is most often associated with lymphoproliferative disorders (most commonly Non-Hodgkin lymphomas such as splenic marginal zone lymphoma) and monoclonal gammopathy of undetermined significance, among other conditions.1,2
In AAE-C1INH, the deficiency of C1 esterase inhibitor (C1INH) leads to an excessive production of bradykinin, which in turn increases vascular permeability leading to angioedema.2 -4 The treatment for this condition focuses on treating the underlying condition and medications such as plasma-derived C1INHs, kallikrein inhibitors, and bradykinin receptor antagonists.1,4 In the upper airway, severe cases may cause potentially fatal asphyxiation.5 -7 In some cases, AAE-C1INH may be the first clinical sign of an occult lymphoproliferative disorder, thus necessitating a hematologic work-up.4,6,8 Treatment of the underlying malignancy may help to improve the condition of the patients in some instances.4,6,8 -10
Herein, we present a 45-year-old female with recurrent angioedema presenting as gastrointestinal complaints who was found to have extranodal marginal zone lymphoma.
Case Presentation
A 45-year-old female from Panama presented with complaints of recurrent episodes of abdominal pain, bloating, and distension for 3 years prior to diagnosis. She was evaluated by multiple GI specialists in her home country and underwent extensive testing including blood tests, endoscopies, imaging studies, and laparoscopic evaluation without receiving a specific diagnosis, except for elevated liver enzymes. A year prior to her diagnosis, she started to complain of episodes of pain and swelling in her neck, shoulders, feet, and both hands mostly in the knuckles, associated with joint stiffness. She denied any rash, hives, or pruritus. This prompted further evaluation by rheumatology, internal medicine, and orthopedic specialists which only yielded a weakly positive rheumatoid factor. Steroids provided only temporary relief. Due to persistence of symptoms, she was evaluated by an immunology specialist. Further testing showed decreased complement levels suggesting the possibility of acquired angioedema. Given the lack of access to specific treatment in her home country, she decided to travel to the United States for further management.
After establishing care at our institution, the patient underwent additional blood test that showed a mildly elevated rheumatoid factor 36 IU/mL (<15 IU/mL), C4 <3 (14-40 mg/dL), C3 within normal limits, C1q 7 (12-22 mg/dL). Her C1-INH levels were low with functional activity at 21% (normal ≥67%) and C1-INH antigen at 4 mg/dL (19-37 mg/dL). She tested negative for anti-C1INH antibodies. Functional C1-inhibitor activity was measured using a chromogenic assay, antigenic C1-INH and C1q levels were determined via nephelometry, and anti-C1-INH antibodies were assessed using an ELISA. The laboratory findings of low levels of C4, and C1q, as well as low C1-INH function and antigen confirmed the diagnosis of acquired angioedema due to C1IHN deficiency (AAE-C1INH). Given the established association between AAE-C1INH and hematologic malignancies, she underwent further workup.
Flow cytometry of peripheral blood showed a CD5 negative, CD10 negative B-cell lymphoproliferative disorder with restricted kappa light chain expression. A bone marrow aspirate and biopsy revealed 40% involvement with low grade B-cell lymphoma, which was consistent with extra nodal marginal zone lymphoma (Figure 1). The molecular testing was negative for the MYD88 mutation, and the PET-CT scan revealed possible splenic involvement. Thus, the diagnosis could be considered splenic marginal zone lymphoma.

The bone marrow biopsy showed nodular proliferation of small lymphocytes (A, H&E ×20), which were positive for CD20 by immunostaining (B, ×20). Flow cytometry analysis or peripheral blood identified a monoclonal B-cell population, which was positive for CD19 and CD20 with kappa light restriction and negative for CD5 or CD10 (flow dot plots C-E).
The patient received rituximab for 4 weeks and her angioedema symptoms resolved completely, C1q normalized (12 mg/dL), and C1INH functional levels started to increase (49%), before reaching complete normalization (68%). These changes are reflected in Figures 2 and 3. Subsequent PET-CT scan confirmed complete resolution of the splenic lesion. A post treatment bone marrow aspirate and biopsy showed a decrease involvement by the lymphoma to 10% of the marrow space. Therefore, rituximab consolidation therapy every 8 weeks for 4 doses commenced.

Complement markers levels after rituximab therapy.

C1-INH functional levels after rituximab therapy.
Following consolidation therapy, a bone marrow aspirate and biopsy revealed no obvious involvement with lymphoma except for a small suspicious (0.1%) kappa-restricted B cell clone. She continued to be symptom-free since therapy initiation and is on surveillance with laboratory checks every 6 months. She was followed for 1 year after finishing consolidation therapy at the time of submitting this report.
Discussion
This case demonstrates the difficulties diagnosing AAE-C1INH and the importance of understanding its association with underlying lymphoproliferative disorders. The 45-year-old patient presented with abdominal pain and peripheral swelling that led a multidisciplinary team to discover findings consistent with AAE-C1INH. Further examinations uncovered an extranodal marginal zone lymphoma (EMZL) with involvement of the bone marrow and possible spleen involvement. Inducing lymphoma into a remission led to the disappearance of angioedema symptoms and correction of C1-INH functional levels.
Angioedema is divided into 2 main categories based on its pathophysiology as histamine-mediated (mast cell-mediated) or bradykinin-mediated, with histamine-mediated being the most frequent type. Bradykinin-mediated angioedema further divides into hereditary (HAE) or acquired (AAE) C1INH deficiency. 2
AAE-C1INH constitutes a rare form of angioedema which appears approximately 10 times less frequently than hereditary angioedema due to C1-inhibitor deficiency (HAE-C1INH) with a reported incidence of approximately 1:500 000.1,4,6,11,12
The most common symptoms of AAE-C1INH include mucosal and soft tissue non-pruritic swelling, mostly located in the face, abdomen, extremities, upper airways (potentially life-threatening), and genitals, which generally lasts under a week. Compared to histaminergic which is a pruritic swelling that lasts for hours. Pathophysiology relies on the role of C1IHN as a serine protease inhibitor that manages multiple inflammatory pathways in the body. Within the complement system C1INH acts to inhibit C1r and C1s components of the classical component pathway while it also inactivates mannan-binding lectin serine protease-2 (MASP-2) in the lectin pathway which activates C4 and C2. C1INH deficiency leads to excessive consumption of complement elements leading to the observed laboratory abnormalities in this condition. The contact-kinin system depends on C1INH to deactivate kallikrein together with factor XII. The unregulated activity of these enzymes creates higher bradykinin levels which then cause swelling through increased vascular permeability.10,13 -16
Multiple pathophysiology mechanisms of AAE-C1INH remain unknown even though several theories attempt to explain them. One theory proposes that antibodies against C1INH generated by abnormal B-cell proliferation could be the underlying mechanism behind the disease.8,11,17 Moreover, multiple studies demonstrate that significant patient populations have C1INH antibodies.6,12 -14,17 -19 Another theory suggests that abnormal tumor tissue or autoantibodies could lead to over activation of the classical complement pathway and subsequent increased consumption of C1INH. Based on the above, it has been suggested that AAE-C1INH may present as either Type I or Type II forms. The conditions often develop alongside lymphoproliferative diseases but do not necessarily display detectable anti-C1-INH antibodies (Type I) or show anti-C1-INH autoantibodies with or without overt malignancy (Type II).5,19 However, this classification is not universally accepted, and overlapping clinical features may complicate its practical application (p. 20).4,7,19 The authors believe that this division lacks practical application because the distinction between the 2 subtypes does not change treatment plans or survival outlook. Additional research is required to explain AAE-C1INH mechanisms better along with determining if this classification system provides useful clinical benefits.
The diagnostic criteria for AAE-C1INH include the following: recurrent episodes of angioedema without urticaria, decreased levels of C1q, C1INH antigen, and/or functional activity less than 50%, onset of symptoms after age 40 years, and absence of familial swelling episodes. Autoantibodies to C1INH, although not always present, help confirm the diagnosis. In contrast, patients with HAE-C1INH develop angioedema episodes early in life, have a positive family history of angioedema, SERPING1 gene mutations, and normal C1q levels.5,8,11 -19 It is important to mention that, despite the fact that AAE-C1INH mimics the symptoms of HAE-C1INH, the former is not caused by genetic mutations, reason why AAE is classified as a phenocopy of HAE in the Inborn Errors of Immunity (IEI) classification by the International Union of Immunological Societies (IUIS). 20
Even though the diagnostic criteria are clearly defined, AAE-C1INH presents a major management problem because the diagnosis is often delayed.6,11,13 The diagnostic process lasted almost 3 years for our patient which matched the results of previous studies.3,11,13 Bork et al 12 and Baeza et al 6 reported a 1-year gap between the onset of symptoms and the AAE diagnosis, while Polai et al 7 described a 5-year delay. This demonstrates the variability in diagnosis delay, probably driven by particular differences inherent to each study population. In addition, multiple studies reported improvement of symptoms and complement levels with early diagnosis and treatment of AAE and its underlying conditions, stressing the need for a timely diagnosis10,17
The classic age of diagnosis for patients with AAE-C1INH is around 60 years.7,11,13,17 -19 However, recent studies have reported cases among younger patients similar to our case.4,6 Furthermore, a study conducted by Baeza et al 6 reported 4 patients diagnosed before their 40s, with the youngest one being 24 years old. Therefore, AAE-C1INH should be considered a potential diagnosis even among young patients, especially if the condition presents classic features such as complement abnormalities alongside non-histaminergic angioedema. 6
The established association between AAE-C1INH and a wide array of underlying conditions, especially malignancies, prompts a timely and thorough evaluation of every patient by a multidisciplinary team involving immunology specialists along with hematology and oncology experts.14,17 -19 A complete diagnostic workup for patients with AAE-C1INH should contain complement tests (C1INH antigen and function, C4, and C1q levels) in combination with complete blood count with differential, peripheral blood flow cytometry, serum monoclonal studies as well as a bone marrow aspirate and biopsy and appropriate imaging studies.3,8
A focused review of major published cohort and case series studies demonstrating the most frequently encountered AAE-C1INH-related diseases was included (Table 1). The studies were identified primarily through targeted literature searches in PubMed and selected based on the sample size, clinical relevance, and available data on associated diagnoses. We examined 11 studies with a total of 372 patients from Europe and the United States. The most common AAE-C1INH-associated condition was non-Hodgkin lymphoma (NHL) in 141 patients (38.2%), particularly splenic marginal zone lymphoma (58 cases, 15.6%). Monoclonal gammopathy of uncertain significance was diagnosed in 103 patients (27.7%). Thirty-four cases (9.1%) exhibited autoimmune conditions including rheumatoid arthritis, Sjögren’s syndrome, and antiphospholipid syndrome. Nine patients (2.4%) had non-hematological malignancies, which included breast, prostate, bronchial, gastric, and urinary bladder adenocarcinomas. It is worth noting that 56 patients (15.1%) did not have any identifiable underlying diseases, which reinforces AAE-C1INH’s complex nature and the requirement for additional research into its pathophysiology.8,10,11,13,14,17 -19
Largest Cohorts of Patients With Acquired Angioedema With C1-inhibitor Deficiency and Associated Conditions Reported in the Literature.
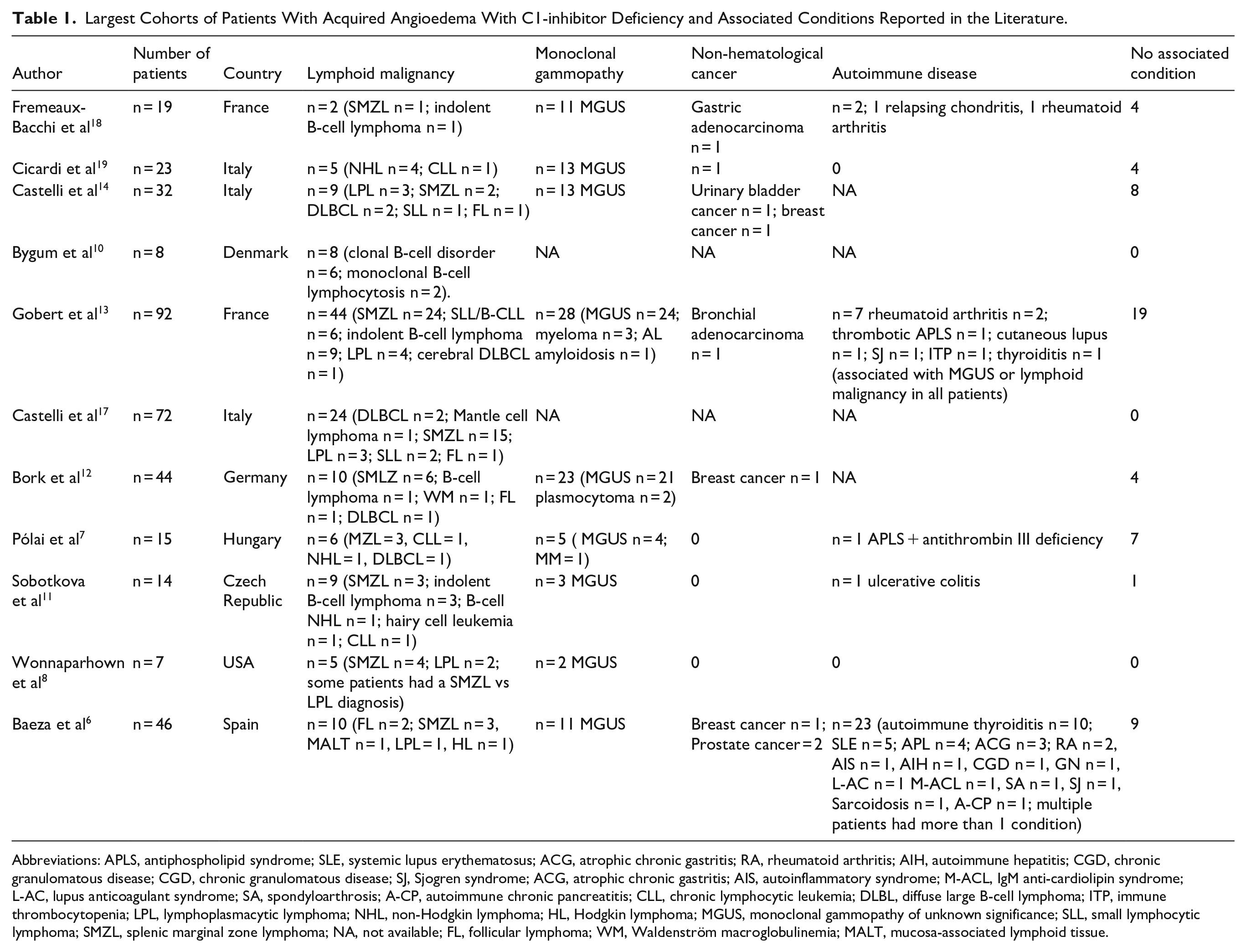
Abbreviations: APLS, antiphospholipid syndrome; SLE, systemic lupus erythematosus; ACG, atrophic chronic gastritis; RA, rheumatoid arthritis; AIH, autoimmune hepatitis; CGD, chronic granulomatous disease; CGD, chronic granulomatous disease; SJ, Sjogren syndrome; ACG, atrophic chronic gastritis; AIS, autoinflammatory syndrome; M-ACL, IgM anti-cardiolipin syndrome; L-AC, lupus anticoagulant syndrome; SA, spondyloarthrosis; A-CP, autoimmune chronic pancreatitis; CLL, chronic lymphocytic leukemia; DLBL, diffuse large B-cell lymphoma; ITP, immune thrombocytopenia; LPL, lymphoplasmacytic lymphoma; NHL, non-Hodgkin lymphoma; HL, Hodgkin lymphoma; MGUS, monoclonal gammopathy of unknown significance; SLL, small lymphocytic lymphoma; SMZL, splenic marginal zone lymphoma; NA, not available; FL, follicular lymphoma; WM, Waldenström macroglobulinemia; MALT, mucosa-associated lymphoid tissue.
Even among patients with AAE-C1INH and no identifiable underlying conditions, experts recommend yearly re-examinations for lymphoproliferative disorders.4,19 In such cases where AAE-C1INH lacks specific treatment approval, the therapy draws from HAE-C1INH-specific medications. Treatment is tailored to treat either acute attacks or for long-term prophylaxis.
Options available to treat acute attacks include:
- Plasma-derived C1INH concentrate (pdC1-INH), Kallikrein inhibitors, such as ecallantide, and B2 receptor antagonists, like icatibant.4,6,10 -13,15,16,18,21
While options for long-term prophylaxis include:
- Antifibrinolytics, such as tranexamic acid, and attenuated androgens, like danazol or stanozolol.4,6,10 -13,15,18,21
Management of the associated condition, if known, has shown consistent benefit for AAE-C1INH patients.5,8,10,11,17 The patient under examination showed complete symptom resolution after treating the lymphoma, mirroring results of several studies4,11,13 In the study by Castelli et al, 17 all patients showed significant improvement of angioedema symptoms after treatment with chemotherapy. In their retrospective study, Zingale et al 22 reported several instances of improvement in angioedema symptoms following treatment of the underlying condition. According to Polai et al, 7 rituximab was the most used agent to treat the underlying diagnosis. It is worth noting that the primary objective of the treatment in our case was to eradicate the underlying lymphoma to improve symptoms, rather than fully correcting complement levels (Figures 2 and 3).
In conclusion, confirming acquired angioedema requires specialized testing of completement and could be challenging thus early referrals to an immunologist is warranted in suspected cases. Additionally, suspicion for lymphoproliferative disorders should be high in cases of acquired angioedema. CBC with differentia, flow cytometry on the peripheral blood, monoclonal studies, bone marrow aspirate, and biopsy as well as appropriate imaging studies is warranted in such cases. Future research should investigate the mechanism of action in relation to the association of AAE-C1INH with certain lymphomas such as splenic marginal zone lymphoma.
Key Clinical Message
Acquired angioedema (AAE) is tightly linked to lymphoproliferative disorders, particularly marginal zone lymphoma (MZL). Assessment of complement abnormalities (low C1q, C4) aid diagnosis. Early recognition and a multidisciplinary approach are essential for management and treating of underlying causes of AAE.
Footnotes
Acknowledgements
We would like to thank Dr. Alexei Gonzalez Estrada, an immunology expert at Mayo Clinic Arizona, for his valuable review and insightful feedback on this study.
Ethical Considerations
Our institution does not require ethical approval to report individual cases. Additionally, the study adhered to institutional and ethical guidelines for the use of de-identified patient information, which do not require explicit consent for publication.
Consent to Participate
Informed consent for patient information to be published in this article was not obtained because the data was anonymized and aggregated, ensuring that individual patients could not be identified.
Author Contributions
Juan Carlos Cardenas Rosales: Conceptualization, investigation, methodology, and writing – original draft. Ahmad Ridwan: Conceptualization, investigation, methodology, and writing – original draft. Carlos Ruiz-Orasma: Writing – writing review and editing, conceptualization, visualization, and literature review. Santiago F. Galeano-Lovera: Investigation, data curation, resources, writing – review and editing, and conceptualization. Jhonny Perusina: Data curation, resources, and writing – review and editing. Jacqueline D. Squire: Formal analysis, supervision, and writing – review and editing. Liuyan Jiang: Investigation, data curation, visualization, and writing – review and editing. Muhamad Alhaj Moustafa: Resources, methodology, supervision – review and editing, and conceptualization. Salma Iftikhar: Methodology, literature review, and writing – review and editing. Dana M. Harris: Literature review, writing – review and editing, and conceptualization. Bala Munipalli: Supervision, literature review, methodology, and writing – review and editing.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.