
Abstract
Alzheimer’s disease (AD) is the most prevalent age-related neurodegenerative disease which is mainly caused by aggregated protein plaques in degenerating neurons of the brain. These aggregated protein plaques are mainly consisting of amyloid β (Aβ) fibrils and neurofibrillary tangles (NFTs) of phosphorylated tau protein. Even though the transgenic murine models can recapitulate some of the AD phenotypes, they are not the human cell models of AD. Recent breakthrough in somatic cell reprogramming made it available to use induced pluripotent stem cells (iPSCs) for patientspecific disease modeling and autologous transplantation therapy. Human iPSCs provide alternative ways to obtain specific human brain cells of AD patients to study the molecular mechanisms and therapeutic approaches for familial and sporadic forms of AD. After differentiation into neuronal cells, iPSCs have enabled the investigation of the complex aetiology and timescale over which AD develops in human brain. Here, we first go over the pathological process of and transgenic models of AD. Then we discuss the application of iPSC for disease model and cell transplantation. At last the challenges and future applications of iPSCs for AD will be summarized to propose cell-based approaches for the treatment of this devastating disorder.
Keywords
1 Introduction
Alzheimer’s disease (AD) is clinically characterized by memory loss and cognitive impairment, and affects the living quality of patients. AD is increasing rapidly within our ageing population. Recent studies reported that 3%~5% of population over the age of 65 is possibly suffered from AD with an estimated 5~7 million new cases of AD each year. Pathologically AD is characterized by deposition of extracellular amyloid plaque and intracellular neurofibrillary tangle (NFT) [1, 2]. Past studies have shown that AD is mainly caused by degeneration of neurons throughout the brain and particularly in the areas of basal forebrain, hippocampus and cortical brain. It is reported that the majority of AD cases are late onset and sporadic, caused by multiple factors which interacting with environmental exposures. Only less than 5% of AD cases are familial and are caused by genetic mutations in the disease genes of APP, PSEN1, PSEN2 and other related genes [3 –5].
Currently there is no effective treatment to stop the disease progression of AD. However the cholinesterase inhibitors (tacrine, donepezil, rivastigmine, or galantamine) and an N-methyl-D-aspartate (NMDA) receptor antagonist (Meman tine) are available for the treatment of AD. Some anti-Aβ monoclonal antibodies, γ-or β-secretase inhibitors were also developed on clinical trials to inhibit the formation and aggregation of amyloid β (Aβ) plaques. In addition, some small molecules and growth factors which can decrease Aβ deposit are also in the ongoing clinical trials [6 –9]. There is an urgent need to develop innovative therapies for AD.
AD is mainly affecting the central nervous system (CNS) of the patients. It is impossible to obtain brain samples from the patients before their death to deeply study the pathological process and treatments of AD. Over the last two decades, the potential use of stem cells to treat the memory dysfunction and cognitive impairment has received growing attention. Specifically, neural stem cell transplantation as a treatment for AD offers a novel approach with tremendous therapeutic potential. Recent advance in somatic reprogramming has generated induced pluripotent stem cells (iPSCs) by over-expressing the KLF4, SOX2, c-MYC and KLF4 genes in somatic fibroblasts or blood cells [10, 11]. The iPSCs are pluripotent and can be differentiated to neural stem cells or neurons for modeling the pathological mechanisms, drug screening and transplantation therapy of the neurodegenerative diseases. In the in vitro studies we can also use the iPSCs to identify new molecular targets to recapitulate the genetic background of the individual from iPSC models. Here we discuss the recent application studies of iPSCs in modeling and treatment of AD.
2 Experimental and clinical studies to uncover the molecular pathological mechanisms of AD
In the past 30 years of studies to investigate the pathological mechanisms of AD by advanced molecular and cellular technology, the major discovery is that in degenerating neurons the Aβ plaques and NFTs were identified as the pathological hallmarks of AD [12]. In the development of AD, cholinergic neurons and synapses are first affected and neurons eventually degenerate to lead to the formation of Aβ deposit and tau tangles in different brain regions. Other studies indicated that glial cells are also involved. Familial AD cases are mainly caused by the mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1 or PS1) and presenilin 2 (PSEN2 or PS2). Sporadic AD patients are usually accredited by multiple factors including environmental, living conditions and other risk factors.
The APP gene was the first AD related gene described in 1991. Later, AD was also found to be associated with mutated genes at early-onset (PSEN1/2). The other AD-associated genes include late-onset genes (apolipoprotein E, ApoE), tau and several potential risk genes [13, 14]. Studies showed that the Aβ peptide associated with phosphorylated tau protein induced impairments of neural cells and synaptic plasticity, indicating tau protein is required for the neurotoxic effects of Aβ deposits. The hyperphosphorylated tau and Aβ peptide are found to be directly interacted to induce aggregation and hyperphosphorylation of tau protein [15 –18]. These Aβ plaques and tau protein-related neurofibrillary tangles spread in all areas of brain and induce the cytotoxicity and cell death of the neurons. The memory and language dysfunctions of patients are caused by increased soluble and insoluble Aβ peptides, which are produced from the sequential proteolytic processing of APP. The aggregated Aβ plaque and tau-related NFTs are formed in the brain to induce extensive neuronal degeneration and neuronal cell death. APP is normally cleaved by β-or γ-secretase in the cell membranes and is highly expressed in the CNS to regulate synapse formation, neurogenesis, axonal transport, cell signaling and plasticity. When APP is sequentially cleaved first by β- and then γ-secretase, the Aβ peptide fragments of 40 or 42 amino acid (Aβ40 and Aβ42) are produced. These Aβ peptides eventually formed Aβ-oligomers or Aβ-polymers to aggregate in the neural cells to form Aβ plagues [19]. PSEN1 and PSEN2 are transmembrane protein components of the γ-secretase enzyme which cleaves multiple substrates including APP. In addition to γ-secretase activity, PSEN1 or PSEN2 is also involved in the regulation of the endosome/lysosome pathway and autophagy [2]. The NFTs are mainly composed of tau aggregates. In the process of NFT formation, tau protein is first phosphorylated, and the phosphorylated tau proteins then aggregate to form highly ordered fibrils and the NFTs [20, 21]. Because of the deposits of Aβ plagues and NFTs in the brains of Alzheimer’s patients, the neural cells shrink and progressively lead to apoptosis. Such programmed cell death occurs first in the brain regions responsible for memory and language, and ultimately spreads to the entire brain. In the apoptotic process of neuronal cells, cholinergic neurons and neuronal synapses are first affected and gradually degenerate to induce brain shrink. The neurons of many brain regions are then affected and have amyloid plaques and neurofibrillary tangles [22 –24].
3 Current transgenic animal models to study AD
To understand the molecular pathogenesis and search for the therapeutic targets of AD, different animal models have been developed. Most transgenic animal models for AD are based on the disease genes identified from familial AD. The first successful APP transgenic mice were generated by over-expressing the APP gene and demonstrated Aβ deposit at early age similar to those of human AD. These mice eventually developed AD-like Aβ deposits in the cortex and hippocampus of their brains by 9~12 months after birth, but failed to develop tau-related NFTs like seen in brains of AD patients [25, 26]. The transgenic mice over-expressing PSEN1 were reported to have some related neuronal loss and increased Aβ42 in the cortex and hippocampus of the mouse brains, but abnormal pathological Aβ deposits of AD were not observed [27]. To overcome the shortages of the single genetransgenic mouse model, a double transgenic mouse model was generated from a cross between the transgenic APP line (Tg2576) and transgenic PSEN1 line (with M146L mutation), and was shown to develop large numbers of fibrillary Aβ deposits in cerebral cortex and hippocampus much earlier than the singly transgenic Tg2576 mice. The neuron loss was seen to be more severe in these double transgenic mice with both mutations of APP and PSEN1 (APPS751L/PS1M146L) and Aβ plaques in cerebral cortex and hippocampus were able to be seen compared to that in the control mice. The double transgenic mice also showed a selectively increase in Aβ42 in their brains. Importantly, a lot of synaptophysinimmunoreactive presynaptic boutons (SIPBs) were seen in the dentate gyrus (SM), stratum lucidum of area CA3 (SL), and stratum radiatum of area CA1-2 (SR) in these double transgenic mice [28]. This study supported the role of PSEN1 mutations to induce neurodegeneration of the nervous systems. In order to further explore the molecular pathological process induced by combined genetic mutations of three genes, subsequent triple transgenic mouse model harboring APP (Swe), PSEN1 (M146V), and tau (P301L) transgenes was created and was found to have AD-relevant pathological features in the brain regions of these triple transgenic mice. These triple transgenic mice developed extracellular Aβ deposits and NFTs. Importantly the triple transgenic mice showed impaired synaptic plasticity including long-term potentiation, synaptic dysfunction and cognitive function before plaque and tangle pathology. These three-gene combined AD mice further supported the relationships between Aβ, tau tangles, providing a valuable model for evaluating potential AD therapeutics [29, 30]. However the progressive neuronal degeneration of cortex, hippocampus and other specific neocortical regions in the human AD brain is not evident in most of the transgenic mouse models, and this is a major limitation of these murine models. Currently there is no mouse model that can fully reproduce the features of pathological progression of majority of sporadic or late-onset AD cases. To investigate effects of the intracellular Aβ (iAβ) accumulation on the molecular neuropathology, the rat transgenic AD models were also generated by expressing APP and PSEN1, in which the impact of iAβ on brain sectional areas of the Golgi apparatus, lysosomes and lipofuscin bodies was analyzed [31, 32]. A recent study developed a transgenic rat line (McGill-R-Thy1-APP) to express the mutant human APP carrying double mutations. These transgenic rats have an intraneuronal Aβ accumulation at about 1 week after birth, which is widespread throughout different cortical areas and the hippocampus (CA1, CA2, CA3, and dentate gyrus). Homozygous transgenic animals eventually produce extracellular Aβ deposits and by 6 months of age, the dense Aβ plaques are able to be detected and associated with glial activation and surrounding dystrophic neurites. These rats were found to have impairment of cognition as early as at 3 months of age and develop extracellular Aβ deposits by 6 months of age [33]. Nevertheless few rat models have been reported to develop both intracellular and extracellular Aβ accumulation in the degenerating neurons and behavior dysfunction including cognitive deficits.
4 Patient-derived iPSCs for AD Modeling
Since transgenic mouse or rat models cannot fully recapitulate the pathological characteristics of Aβ plaques and the tau tangles, and also the phenotypes of memory and cognitive dysfunctions, recent iPSC technology provided us the great opportunity to use the human cell models to study the pathological process of AD and screening therapeutic agents for treatment of AD. As we cannot perform the brain analysis of AD patients, these iPSCs can be used to study pathological mechanisms of the diseased neural cells [34].
4.1 Using iPSCs with genetic mutations to model familial AD
Most available iPSCs are from AD patients who have genetic mutations in the associated genes. These iPSCs were used to investigate the neural phenotypes and electrophysiologically activities, which are from patients diagnosed with either familial AD mutations in APP or PSEN1, or from patients with the genomic duplication of the APP gene which results in increased Aβ deposit. Most studies have shown an increased total Aβ protein or the specific Aβ42, resulting in an increase of the Aβ42/40 ratio. An increase in the aggregated Aβ42 or in the Aβ42/40 ratio accelerates the disease progression through the production of toxic oligomeric or fibric aggregated Aβ to form Aβ deposits in the iPSC-derived neural cells [35]. Koch et al. reported that PS1-L166P mutant human pluripotent stem cells were differentiated to neural stem cells and neurons which express the neuron-specific APP (695) splice variant, BACE1, and all members of the γ-secretase complex [36]. These iPSC-derived neurons also exhibit an increase in Aβ secretion and respond to the therapeutic effect by anti-amyloidogenic compounds, such as γ-secretase inhibitors, indicating the usefulness of iPSCs as human cellular models to study the molecular mechanisms of AD. Another study reported iPSCs from familial AD patients with APP-E693 deletion and the iPSCs were differentiated into neural cells [37]. In these iPSC-derived neurons and astrocytes, Aβ oligomers were observed to accumulate and lead to toxicity to cellular endoplasmic reticulum (ER) and lysosomes. A subsequent study reported that iPSCs from patients harboring the APP mutation (V717I) were induced to differentiate to forebrain neurons [38]. The important finding of this iPSC model is that mutation-containing iPSCs require longer time to mature to neuronal fates. The APP expression and the levels of Aβ were observed to increase dramatically in the mutation-containing neurons compared to that of normal iPSC-derived neurons. In addition, this mutation was found to alter the initial cleavage site of γ-secretase, leading to an increase of specific Aβ42, Aβ38 and phosphorylated tau in iPSC-derived neurons with the APP-V717I mutation. In the pathogenesis of AD, the AD-associated PSEN1 mutation (L166P) caused a partial functional loss of γ-secretase, resulting in the decreased Aβ40 and an increased Aβ42/40 ratio. To examine if the PSEN1 mutations (A246E, H163R, and M146L) affect the effectiveness of γ-secretase inhibitor and γ-secretase modulator (GSM), iPSC-differentiated neurons were compared between patients carrying 3 different PSEN1 mutations and normal control individuals. As a result, mutant PSEN1 neurons exhibited a significant elevated Aβ42/40 ratio as compared with the control neurons. Importantly, treating the iPSC-derived neurons with a potent antiinflammatory drug GSM identified new biomarkers that differs from all previous cell types and animals tested. This new biomarker panel consisted of a reduction in Aβ42, Aβ40 and Aβ38 and in the Aβ42/40 ratio, with no change in the total Aβ levels. This study provided a unique signature that will more accurately reflect drug response in human AD patients and in cerebrospinal fluid biomarker changes observed during GSM treatment [39].
To further validate the reliability of iPSC with genetic mutations to model pathogenesis of AD, the genetic mutations can be corrected to examine the phenotypes and function of the iPSC-derived neurons. The availability of CRIPSR/Cas9 gene editing has generated mutation-corrected iPSCs for AD modeling. This gene editing has been used to create different iPSC lines from AD models with mutations in APP. The gene-corrected iPSCs from AD patients carrying mutations were also generated to understand the phenotype recovery of these iPSCs. Using episomal plasmids expressing hOCT4, hSOX2, hKLF4, hL-MYC, hLIN28 and a short hairpin against TP53, Li et al. showed that induced pluripotent stem cells derived from a 48-year-old woman carry a heterozygous mutation in exon 4 of the PSEN1 gene, which causes a change in amino acid A79V (c.236 C to T) [40]. This iPSC cell line was reported as a bona fide iPSC line with normal karyotype. Later on, the gene-corrected iPSC line was generated by the CRISPR/Cas9 system to replace the point mutation T with the wild-type nucleotide C. iPSC-derived basal forebrain cholinergic neurons (BFCN) with the PSEN2 N141I mutation displayed an increased Aβ42/40 ratio. Neurons derived from PSEN2 N141I iPSCs were found to have fewer numbers of spikes in response to a depolarizing current. The height of the electrophysiological action potential was also significantly decreased in BFCNs with PSEN2 N141I mutation. CRISPR/Cas9 correction of this PSEN2 point mutation eliminated the electrophysiological deficit of increased Aβ42/40 ratio, restoring the number and height of spikes to the levels of iPSCs from control individuals. The genome editing analysis confirms the recovery of mutation-related changes induced by Aβ42/40 ratio [41].
Most recently a CRISPR/Cas9 edited APP-C-terminus showed that attenuating APP-β-cleavage and Aβ production were decreased while the neuroprotective APP-α-cleavage was up-regulated in APP-corrected iPSC-derived neurons. The cellular and secreted APP products were examined in APP edited human iPSC-derived neurons. C-terminus selective-APP editing in both WT and mutant APP lines revealed that increased sAPPα in both WT and London lines, suggesting upregulation of the neuroprotective α-cleavage pathway. ELISAs and western blot showed attenuated secretion of Aβ40/42 and sAPPβ confirming inhibition of the amyloidogenic pathway in these neurons. Genomic deep sequencing showed efficient editing of human APP by the sgRNA, with truncation of the last 36 amino acid in human embryonic stem cells [42].
iPSC models for understanding of the disease pathology are mostly used on neuronal cells, however the astrocytes have also been reported to contribute to AD. One study generated functional astrocytes from iPSCs of AD patients with PSEN1 ΔE9 mutation and gene-corrected patients. It was found that AD patient-derived astrocytes showed characteristical pathology of AD, such as increased Aβ production, altered cytokine release, and disturbance of Ca2+ homeostasis. Furthermore, AD astrocytes were observed to have increased oxidative stress and reduced lactate secretion, and compromised function of astrocytes, compared to that in healthy neurons. This indicated that astrocytes are playing important role in AD pathology and suggested the strength of iPSC-derived astrocyte models for disease mechanisms of AD [43].
4.2 Using iPSC‑Derived Neurons to model sporadic AD
Sporadic AD (sAD) accounts for the majority of the AD cases. A variety of studies have generated iPSCs from patients with sporadic AD. Some studies compared the neuron phenotypes of iPSCs from familial AD with genetic mutation, sporadic AD and control individuals. It was found that iPSC-derived neurons from the APP duplication patients and sporadic patient exhibited significantly higher levels of the pathological markers Aβ40, phosphorylated tau (Thr 231) and active glycogen synthase kinase-3β (aGSK-3β) althrough all neuron cells exhibited normal electrophysiological activity as compared to controls [44]. A recent study compared the iPSC-derived neurons from patients with early-onset fAD with mutations in the PSEN1 gene, patients with late-onset sAD, and control individuals without dementia. As a result, neurons derived from patients with fAD and patients with sAD exhibited higher levels of extracellularAβ1-40 and Aβ1-42. But, the Aβ1-42/Aβ1-40 ratios were only observed to be significantly increased in patients with fAD. Additionally, the increased levels of active glycogen synthase kinase 3β, a physiological kinase of TAU were found in neurons derived from iPSCs of AD patients. Moreover, elevated sensitivity to oxidative stress was detected in both fAD- and sAD-derived neurons [45].
Most of the studies indicated that the pathological phenotypes of iPSC-derived neurons are similar in patients with fAD and sAD. However a study found that neurons derived from the fAD patient have a higher susceptibility to Aβ1-42 oligomers compared with neurons coming from sAD and healthy individuals [46]. Even though most studies showed that Aβ production was increased in iPSC-derived neurons, few studies have investigated the Aβ clearance for modeling AD. In neurons Aβ degradation is primarily processed through proteolytic degradation and autophagy. In brain neurons of AD patients, the clearance and degradation of Aβ by autophagy is affected to produce increased toxicity of intracellular Aβ. The increased levels of total tau (tTau) and phosphorylated tau (pTau) are playing key roles to increase Aβ aggregation in AD. Several studies indicated that the tau protein-induced pathological process is independent from APP in iPSC models of AD. In addition to tau and ApoE, SORL1 is another susceptible risk factor for sporadic AD (sAD). The pathological effect of these susceptible genes may be difficult to analyze with standard in vitro or in vivo models because of other environmental effects. iPSCs harboring genetic variation in the SORL1 gene may contribute to sAD-related phenotypes in human brains. The neurons from iPSCs carrying SORL1 variants show a reduced SORL1 expression and APP processing were observed by treatment with brain-derived neurotrophic factor (BDNF). Further shRNA knockdown of SORL1 indicated that the BDNF-induced APP processing is dependent on SORL1 expression. This study suggested that the expression variation in SORL1 is modulated by common genetic variants which can contribute to an individual’s risk of developing sAD [47].
In addition to using iPSCs to investigate the intracellular Aβ and tau protein pathogenesis, other molecular mechanisms of AD were also explored by the AD model of iPSCs. In order to model the transcriptome pathology of AD and to analyze the disease phenotype of the ubiquitinproteasome system (UPS), protein expression of AD, and GSK3B, a physiological kinase of tau, were examined in neuronal cells derived from AD-iPSCs. After treatment of neurons from AD-iPSCs with an inhibitor of γ-secretase, phosphorylated tau was down-regulated. Transcriptome analysis of AD-iPS derived neurons revealed significant changes in the expression of genes associated with AD and an AD-related protein interaction network composed of APP, GSK3B and others could be generated using AD-iPSC derived neurons [48].
iPSC derived neuronal models from patients with sAD have also been used to investigate other AD-related phenotypes such as ER stress, mitochondrial dysfunction and oxidative stress [37]. A recent study demonstrated aberrant mitochondrial function in patient-derived cells. The cultured neurons from iPSCs of AD patients produced more reactive oxygen species (ROS), displayed higher levels of DNA damage and increased oxidative phosphorylation chain complexes. However, these pathological phenotypes neither correlated with Aβ nor phosphorylated tau levels. This study therefore highlights the possibility of additional mechanisms of disease pathogenesis in the development of sporadic AD prior to the appearance of amyloid and tau pathology, suggesting novel therapeutic approaches need to be used for treatment of sAD [49]. Some iPSC lines have been generated to model the AD are summarized in Table 1.
Summary of induced pluripotent stem cell models of Alzheimer’s disease.
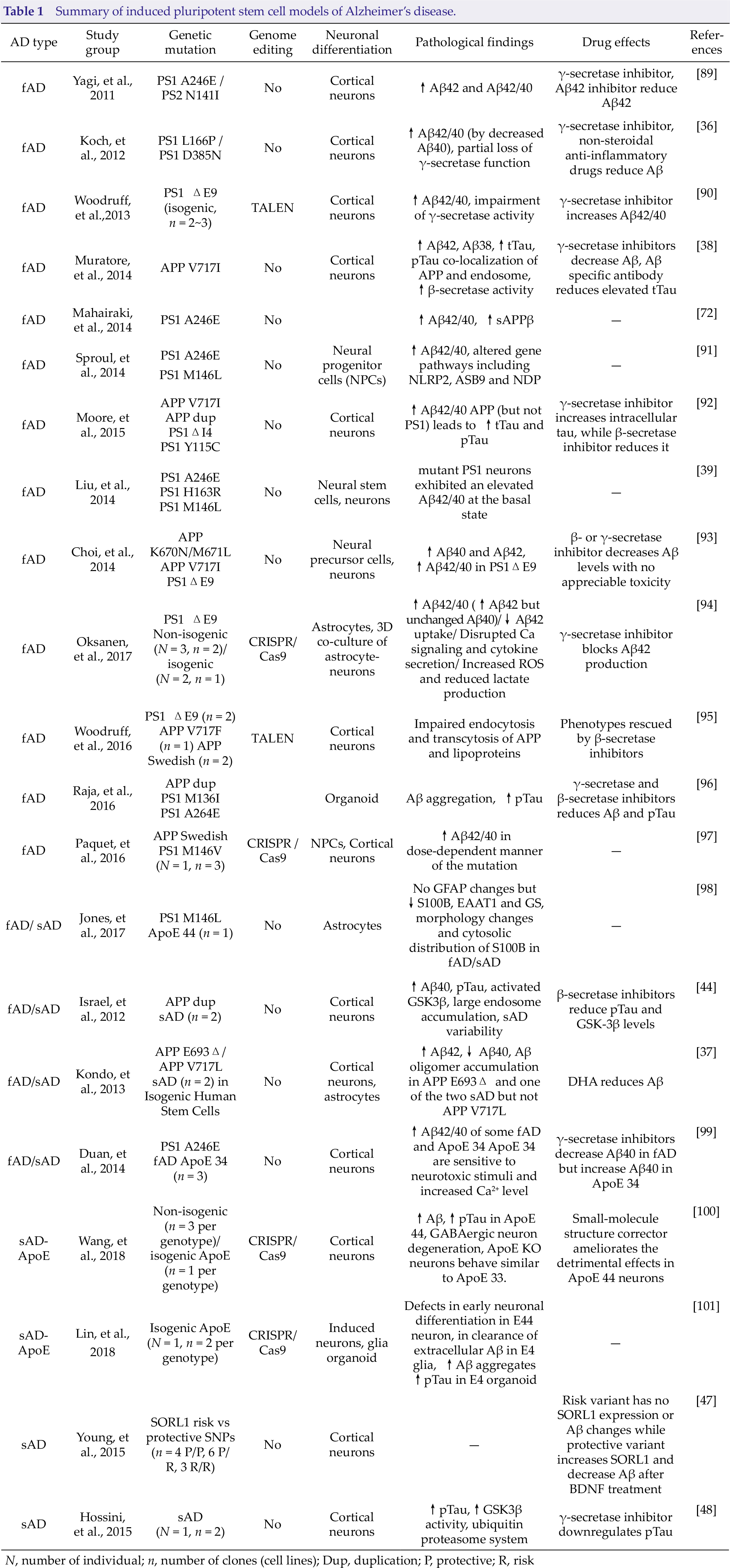
N, number of individual; n, number of clones (cell lines); Dup, duplication; P, protective; R, risk
5 iPSCs-based therapy of AD
Stem cells have been widely investigated as a cell replacement treatment for AD. The therapeutic rational is to take advantage of the transplanted stem cells to replace the degenerated or injured neural cells and release cytokines to activate the endogenous neurogenesis [50, 51]. Currently there are several stem cell sources which have been studied for transplantation therapy of AD. They include embryonic stem cells (ESCs); fetal neural stem cells (NSC); adult stem cells (ASCs) which include hematopoietic stem cells (HSC), mesenchymal stem cells (MSCs) and olfactory ensheathing cells (OECs); somatic reprogrammed cells which include iPSCs and the directly induced neurons (iN) from somatic cell reprogramming [52, 53]. Here we focus on the three kinds of cells as following.
5.1 Fetal neural stem cell therapy
Human fetal neural stem cells (fNSCs) are isolated from the fetal brain of the aborted embryos at 3~4 months of implantation. fNSCs can be cultured in vitro and have self-renewal and neural differentiation potentials. They are able to differentiate to neurons and glial cells in vitro and in vivo [54]. Mouse NSCs (mNSCs) from postnatal day 14 mice were transplanted to hippocampus of the mouse AD model and were shown to improve memory deficits of AD mice. The transplanted NSCs in the hippocampi of the mouse brain were found to survive for at least 5 months and differentiate into neurons and astrocytes [55, 56]. It was also found that transplanted NSC significantly improve synaptic connections and increased neuronal number of host mouse brain, supporting NSCs have some therapeutic roles for AD.
The therapeutic effect of fNSC transplantation on cognitive function was investigated in the APP/PS1 transgenic mouse with age-dependent cognitive deficits. It was shown that the bilaterally transplanted fNSCs into hippocampal regions were able to improve spatial learning and memory function in these mice. However, the pathological Aβ deposits could not be decreased. Transplanted NSCs were able to proliferate, migrate, and differentiate into neurons and glial cells. Importantly the improved cognitive function was related to enhanced long-term potentiation (LTP) and an increase in the expression of neural proteins was observed. These proteins are related to cognitive function and including synaptophysin, protein kinase C ζ subtypes (PKCζ), tyrosine receptor kinase B (TrkB), and BDNF. This study strongly suggested that grafted fNSCs were able to improve cognitive deficits in APP/PS1 transgenic mice [57].
A recent study reported that transplanted fNSCs into bilateral hippocampus of an APP/PS1 double transgenic mouse model of AD improved the recognition and memory deficits of AD mice. The transplanted fNSCs migrated dispersedly in broad brain regions and differentiated into the neurons and astrocytes. The synaptic and nerve fibers of the frontal cortex and hippocampus were found to be significantly increased in the fNSC-treated AD mice, indicating the neuronal connectivity between transplanted human neural stem cell (hNSC) and AD brains. In addition, the Aβ plaques were seen to be reduced in frontal cortex and hippocampus of the mouse brains with transplanted fNSC compared to that in wild-type and PBS-transplanted APP/PS1 mice [40]. To overcome the immune-rejection of human cells transplanted to mouse AD models, one study used murine neural stem cells for transplantation in AD mouse model and found that mNSCs can increase cognition and improve synaptogenesis in 3xTg-AD mice with neuronal loss. They also reported the evidence that transplantation of research grade human central nerve system derived stem cells (HuCNS-SCs) can improve cognition in the mouse models of neurodegeneration. HuCNS-SC cells were found to migrate and differentiate into immature neurons and glia, and significantly increase synaptic and growthassociated markers in both 3xTg-AD and CaM/Tet-DTA mice. However, the pathological Aβ or tau deposits were not reduced by grafted cells in aged 3xTg-AD mice [58].
To increase the survival and neural differentiation of transplanted cells in vivo, some genetic approaches was ever used to modify fNSCs to express cellular growth factors nerve growth factor (NGF) and BDNF to make the fNSC cells expressing NGF or BDNF. Transplantation of fNSCs expressing BDNF and NGF was shown to increase learning and memory function in AD animals [59]. The transplanted fNSC-NGF cells were found to express NGF and improve the learning and memory function in chemically induced AD rats [60]. Since the dysfunction of the cholinergic neurons and decreased activity of choline acetyltransferase (ChAT) are the main cause of cognitive deficits in AD, fNSCs were engineered to express ChAT gene to increase release of ChAT for restoration of cognitive function. Transplantation of fNSC expressing ChAT to the rats with learning deficit, the learning and memory function of the rats were improved with the elevation of ACh levels in cerebrospinal fluid of the rats, suggesting the engineered fNSCs function in vivo [61, 62].
Another type of fNSCs is human olfactory bulb neural stem cell, or OEC, isolated from the olfactory bulb tissue of adult or fetus. These cells are capable of promoting the functions of neurons and glial cells. OECs were able to secrete neurotrophic factor to increase endogenous neurogenesis and the survival of the transplanted cells. Some studies indicated that OECs have been cotransplanted with NSCs or other cells to repair the brain and spinal cord injuries [63 –65]. The expression of choline acetyltransferase was significantly increased in co-transplanted animals than that in transplanted either OECs or NPCs alone to improve cognitive dysfunction in rat model [66]. It was also shown that transplantation of NPCs with OECs to hippocampal regions promoted better recovery of learning and memory of animals lesioned with kainic acids [66]. Most recent genetically modified human olfactory bulb neural stem cells (OBNSCs) have been shown to restore cognitive deficit in rat AD model. These cells were engineered to express human NGF by lentivirus-mediated infection and were transplanted into the hippocampus of AD rats induced with ibotenic acid. The OBNSCs-hNGF cells were found to be differentiated to mature neurons, oligodendrocytes and astrocytes, and alleviate the memory and learning functions of the AD rats [67].
5.2 Embryonic stem cell therapy
Because of limited neural differentiation of the fetal brain derived-fNSCs, the pluripotent ESCs were also studied for transplantation therapy of AD models. One study compared the effects of ESC-derived neuronal precursor cells (ESC-NPCs) and the primed ESC-NPCs (ESC-PNPCs) for treatment of rat AD model [68]. The primed-NPCs were induced from ESCs by addition of Shh to the neural induction medium and were found to have more cholinergic neurons. After transplantation, both ESC-NPCs and ESC-PNPCs improved memory deficits of AD rats. This suggested that the transplantation of mouse ESC-NPCs and/or ESC-PNPCs (commitment to cholinergic cells in vitro) can promote behavioral recovery in a rodent model of AD. To track the migration and in vivo differentiation of ESC-NPCs, mouse ESCs labeled with EGFP were induced into Nestin-positive NSCs in vitro and were then transplanted into the Aβ peptide induced rats. The transplantation of these NPCs into Aβ-injured hippocampus significantly improved the memory function of the Abeta-injured rats 16 weeks after transplantation [69]. Human ESCs were also differentiated into mature BFCNs for the treatment of AD. After transplantation into the basal forebrain of AD model mice, both mouse and human ESC-derived BFCN progenitors differentiate into mature cholinergic neurons in vivo and improved the learning and memory functions of AD model mice [70].
5.3 Induced pluripotent stem cells therapy
Recent advance in somatic cell reprogramming has provided therapeutic potential for neurodegenerative diseases. iPSCs were generated by over-expressing four transcription genes of OCT4, SOX2, c-MYC and KLF4 in skin fibroblast cells, blood cells and urine cells. iPSCs can be derived from patients themselves and can be differentiated into any type of cells including neurons and glial cells. Thus, iPSCs overcome the immune rejections and ethical issues of heterogeneous stem cell transplantation and will have better efficacy for treatment of neurodegenerative diseases including AD. Earlier iPSCs were generated by retroviral or lentiviral vectors to express OCT4, SOX2, c-MYC and KLF4, these exogenous genes and the retroviral or lentiviral vectors may integrate to genome of the iPSCs to cause mutations in iPSCs. Thus, the retroviral or lentiviral-induced iPSCs may not be suitable for cell-based therapy. The transgene-free iPSCs can be derived by episomal plasmids or synthetic mRNA technology [71]. Recently the transgene-free iPSCs were derived from AD patients with PS1 mutation (A246E) using nonintegrating episomal vectors. These iPSCs harboring PS1 gene mutation could be used as human models to study pathogenesis and screen therapeutic drugs for AD. Neurons from the mutant iPSC lines were found to express PS1-A246E mutations and have Aβ deposits as shown in brain of AD patients [72]. The protein-based reprogramming can generate iPSCs more suitable for transplantation therapy because these iPSCs are non-tumorigenic.
The iPSCs were also reported derived from mouse skin fibroblasts by protein extracts of embryonic stem cells. The protein-based mouse iPSCs were able to be differentiated into glial cells and reduced the level of Aβ40, Aβ42, and Aβ deposit in the brains of 5XFAD transgenic mouse model compared with saline-injected control 5XFAD mice after transplantation. The transplanted protein-iPSCs were found to decrease the cognitive dysfunction observed in these mice [73].
However, the iPSC-derived neurons still carry mutations of the APP, PS1 and PS2 genes, these mutated neurons cannot be directly transplanted to AD patients for therapy. To resolve this problem, several approaches have been developed to correct the mutations in the mutant iPSCs by homologous recombination in other studies and then the mutation-corrected neurons can be transplanted to patients for the treatment of AD [42, 74, 75].
In addition to the differentiated neurons of iPSCs, the therapeutic potential of human iPSC-derived macrophage-like cells for AD was also explored. Human iPSCs-derived macrophages were engineered to express neprilysin-2, the Aβ-degrading protease to therapeutically reduce Aβ levels after transplanted to a transgenic mouse AD model. The iPSC-derived macrophage-like myeloid lineage (iPS-ML) cells was made to express the Fc-receptor-fused form of a single chain antibody specific to Aβ and Neprilysin-2 (NEP2) which is a protease with Aβ-degrading activity. In vitro expression of NEP2 enhanced the effect to reduce the level of soluble Aβ oligomer in the culture medium and to alleviate the neurotoxicity of Aβ. To analyze the therapeutic effect of iPS-ML expressing NEP2 (iPS-ML/NEP2) in vivo, the iPS-ML/NEP2 was transplanted to 5XAD mice. A significant reduction in the level of Aβ in the brain interstitial fluid of 5XAD mice was observed. This study suggested that iPS-ML/NEP2 may be a potential therapeutic cell for the treatment of AD [76].
5.4 The molecular mechanisms of stem cell transplantation
The molecular mechanisms stem cell transplantation played to improve the memory and cognition in animal models of AD is not clear, but several aspects of mechanisms have been investigated and the possible mechanisms include cell replacement, release of neurotrophic and neuroprotective factors, endogenous activation of neurogenesis, anti-inflammatory activities, stem cells as carriers to deliver therapeutic proteins to the degenerated regions of brain to decrease Aβ deposits. Neural stem cells can express many neurotrophins including BDNF and NGF [77]. BDNF, glia-derived neurotrophic factor (GDNF) and NGF are the major factors playing regulatory functions in synaptic plasticity. Rats with traumatic brain injury (TBI) were intravenously treated with hMSCs. The extracts from the entire traumatized cerebral hemispheres with grafts 24 h after TBI showed significantly increased expression of NGF, BDNF, and neurotrophin-3 (NT-3) [78]. Stem cells can secrete anti-inflammatory factors such as interleukin-10, an anti-inflammatory cytokine and prostaglandin E2 to inhibit the inflammatory process of AD. Transplantation of NSCs can also influence endogenous neurogenesis of the hosts.
The transplantated NSCs derived from human embryonic stem cells reduced infarct volume and improved behavioral outcome after distal middle cerebral artery occlusion (MCAO) in rats. Jin et al. reported that, transplantation was able to increase neurogenesis expressing doublecortin (Dcx) in subventricular zone (SVZ), but not in contralateral SVZ or dentate gyrus zone (SGZ) of the rat brains 60 days post-transplantation [79]. To determine if MSC stimulates endogenous neurogenesis, MSC was transplanted to the normally restrictive SVZ of mice and was found to increase proliferation and neuronal differentiation of neural progenitors within the SVZ. The proportion of the newborn neurons was increased out of the total proliferating cells [80]. A recent study showed that adipose-derived MSC transplanted into the hippocampi of APP/PS1 transgenic AD mice significantly increased number of BrdU/DCX-stained cells in the in the subventricular zone of the dentate gyrus in the hippocampus, suggesting that MSC transplantation improve the memory and cognitive functions by enhancing the neurogenesis of the APP/PS1 transgenic AD mice [81]. BM-MSCs were shown to increase leukocyte activation and promote leukocyte-endothelial interactions. BM-MSCs were found to significantly inhibit transcriptional activation of NF-kappa B and inhibit DNA binding of NF-kappa B subunits p50 and p65 to putative NF-kappa B DNA binding sites by the direct effects of MSCs on IL-10 and NF-kB [82].
In a study that focused on alleviating amyloid pathology through transplanted NSCs from postnatal mice, the NSCs was modified to express metalloproteinase 9 (MMP9), a protease to degrade aggregated Aβ peptides. Even though some endogenous MMP9 was detected around amyloid plaques in the mouse models, no stem celldelivered MMP9 was found to have impact on the Aβ plaques. This study suggested that the delivering approach may be required to enhance therapeutic efficacy of the transplanted cells [83]. To know how transplanted cells to improved spatial memory, hNSCs were injected into the cerebral lateral ventricles of APPsw-expressing (APPsw) transgenic mice at 13 months of age, it was found that Aβ production was reduced through an Akt/GSK3β-signaling-mediated decrease in BACE1, and the expression of inflammatory mediators was decreased through deactivation of microglia [84].
To increase the production efficiency of iPSC-derived neuronal cells, the automated reprogramming and the pooled selection of polyclonal pluripotent cells have generated high-quality and stable iPSC lines. These lines display less line-toline variation than either manually produced lines or lines produced through automation followed by single-colony subcloning. This platform will enable the application of iPSCs to population-scale biomedical problems including the study of complex genetic diseases and the development of personalized iPSC-based transplantation therapy [85].
6 Clinical studies of stem cell transplantation
The experimental evidence for the safety and efficacy of stem cell-based therapies for AD through animal models strongly supported the approval of several clinical trials. Most of the clinical studies used MSCs from human umbilical cords and bone marrows. In a phase I study conducted in South Korea, human umbilical cord blood mesenchymal stem cells (hUCB-MSCs) were injected to AD patients with mini-mental state examination (MMSE) scores between 10 and 24 to exclude severe AD subjects. The included subjects were divided into 3 groups: a stem cell high-dose group (6.0 × 106 cells), a low-dose group (3.0 × 106 cells) and a treated control group. hUCB-MSCs were injected into the bilateral hippocampus and precuneus directly. Subjects were evaluated after 12 weeks and continue for 24 months by brain computerized tomography (CT), magnetic resonance imaging (MRI), fludeoxyglucose positron emission tomography (FDG-PET), blood samples and clinical evaluation. It was reported that no severe acute or long-term side effects or significant clinical efficacy were found [86, 87]. A multicenter, randomized, placebocontrolled and double-blinded phase I/II study started in 2016, in which forty subjects with probable AD and mixed dementia according to the criteria of the NINCDS-ADRDA were intravenously infused with hUCB-MSCs 8 times at 2-week intervals. This study will compare blood, cerebrospinal fluid (CSF), and neuropsychological assessments among different groups (Clinicaltrials.gov, NTC02672306). Other clinical studies using different injection routes, wellestablished scales, and biomarkers such as amyloid positron emission tomography, are planned for stem cell transplantation to moderate AD patients are ongoing. Recently the clinical cell therapy guidelines for cell-based therapy of neurological diseases were formed to improve the therapeutic effects and standardization of the cell transplantation to the patients [88]. It is believed that advances in iPSCs technology will enable development of stem cell-based therapeutic approaches to increase the clinical use of the stem cell products in AD.
7 Conclusions
iPSCs are pluripotent cells and are able to differentiate to neural lineages to replace the damaged cells for the treatment of AD. Although preclinical studies showed substantial efficacy of stem cell transplantation in AD, few trials showed positive results. Thus, not only do we need to further understand the mechanisms underlining the pathological process of AD and regulating the proliferation, migration, differentiation, survival, and function of iPSCs, but also employ translational models to bridge this gap between the animal studies and clinical trials. Some stem cell sources such as BM-MSC, UC-MSC, and UCB-MSC are easily accessible; however these cells have limited neural differentiation and may induce immune rejections as they are allogenic.
Human ESCs and NSCs have ethical and immune rejection issues which hindered their clinical application in AD therapy. The iPSCs are derived from reprogrammed somatic cells such as fibroblasts, blood or urine cells of patients themselves and overcome the immune rejection and ethical issues on stem cells. Thus patientspecific iPSCs will have invaluable potential for treatment of AD. As the mechanisms of AD are eventually elucidated by animal and human iPSC models, the iPSC-based therapy will become more and more important. The differentiated cell types are major concerns. Some factors such as cell proliferation, differentiation, migration, unity with neural stem cells are also crucial to the actual use of iPSCs. Before the iPSCs are moving to AD patients, several aspects on mutation correction, neural differentiation, purification and long-term survival of iPSCs following transplantation need to be resolved and standardized to make clinicalgrade iPSCs suitable for the AD patients.
Footnotes
Conflict of interests
Dong Han is working as a research assistant for animal studies in the Shandong Molecular Diagnosis & Cell Therapy Biotechnology Corporation.
Financial support
This work was supported by National Natural Science Foundation of China (NSFC 81571241) and Research Start Fund of Shandong University of Traditional Chinese Medicine (2018-220259).