
Abstract
This study aims to investigate the relationship between hepatitis C virus (HCV) NS3/4A and endogenous interferon regulatory factor-3 (IRF-3). The localization of endogenous IRF-3 protein before and after virus infection was analyzed by immunofluorescence assay (IFA). IFA results revealed that the synergistic action of transfection and HCV virus infection could more effectively reduce the nuclear translocation of endogenous IRF-3 in HeLa cells, compared to the activation of Sendai virus infection alone. The highest nuclear translocation of endogenous IRF-3 in transfected HeLa cells occurred at 24 h after Sendai virus infection. Our study was consistent with a published paper, which revealed that HCV NS3/4A protease could suppress the activation of IRF-3 and was indispensable in the transcription of interferon (IFN)-α/β.
Introduction
Viral infections result in the induction of type I interferons (IFNs), which are crucial mediators of antiviral response.1–4 Type I IFNs consist of IFN-β and the large IFN-α family. The expression of IFN-α/β is governed by proteins known as interferon regulatory factors (IRFs). Two members of this family, IRF-3 and IRF-7, are absolutely required for the transcription of IFN-α/β genes.5,6 Quiescent IRF-3 is localized in the cytoplasm. Upon viral infection, IRF-3 is phosphorylated and translocated into the nucleus, where it induces the transcription of IFN-α/β.1,2,5,7
Persistent hepatitis C virus (HCV) infection interacts directly with the host pathway that signals IRF-3 activation. Recent studies have shown that HCV NS3 serine protease could block the phosphorylation and effector function of IRF-3. 7 Furthermore, the disruption of NS3/4A protease function by mutation or a ketoamide peptidomimetic inhibitor relieves this blockade and restores IRF-3 phosphorylation after a cellular challenge with the Sendai virus. Moreover, dominant-negative or constitutively active IRF-3 mutants enhance or suppress HCV RNA replication in hepatoma cells.6–8,9
The IRF-3 gene encodes a 55-kDa protein, which is constitutively expressed in all tissues.2,4,6 To date, most previous studies on IRF-3 have utilized overexpressed IRF-3 proteins as materials by transfecting specific plasmid-containing IRF-3 complementary DNA (cDNA) into mammalian cells.1,5,7 It is necessary to investigate whether the activation of endogenous IRF-3 can be inhibited by the HCV NS3/4A protease. In the present study, the results demonstrate that NS3/4A can prevent endogenous IRF-3 from being translocated into the nucleus.
Materials and methods
Plasmid
Plasmid pSG5-NS3/4A was constructed by other researchers in the same laboratory. The HCV NS3/4A gene was cloned using polymerase chain reaction (PCR) from plasmid pBRTM/HCV1-3011, which includes the full length of the HCV gene. PSG5 was digested sequentially with SmaI and EcoRI. Both digested PSG5 and HCV NS3/4A segments were analyzed by agarose gel electrophoresis. Then, according to molecular size, the expected products were extracted and ligated, and the positive clones were screened by ampicillin and amplified. The recombinant pSG5-NS3/4A was extracted, purified, and identified by restriction endonuclease BamHI and agarose gel electrophoresis.
Cell culture
HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM
Subcellular localization of IRF-3 proteins
Cells were placed on coverslips in a 24-well plate at a concentration of 5 × 104/mL and transfected by plasmids using liposome reagent (FuGENE 6; Roche Molecular Biochemicals) according to reagent instructions. At 24 to 36 h post-transfection, cells were infected with the Sendai virus at a multiplicity of infection (MOI) of 5. At different time points (4, 8, 12, 18, 24, and 36 h post-infection), cells were washed with phosphate buffered saline (PBS) and fixed with ice-cold methanol for 40 min at −20°C. Then, cells were incubated with primary antibodies, including rabbit IRF-3 antibody (Santa Cruz Biotechnology, according to reagent instructions), mouse NS3 monoclonal antibody (MBL, according to reagent instructions), and mice Myc antibody (MBL, according to reagent instructions) for 1 h at room temperature. Next, cells were incubated with secondary antibodies, including anti-rabbit Cy3 (Chemicon International, according to reagent instructions) and anti-mouse fluorescein isothiocyanate (FITC; MBL, according to reagent instructions) for 40 min at room temperature. Coverslips were mounted with antifade reagent containing 4′,6-diamidino-2-phenylindole (Vectashield; Vector Labs, according to reagent instructions). The fluorescence of IRF-3, NS3/4A, and NS3 were analyzed using an Olympus fluorescence microscope with a ×40 objective.
Immunofluorescence staining procedure
The cell-seeded coverslips were first incubated in the primary antibody mixture of rabbit anti-IRF-3 polyclonal antibody and mouse anti-NS3 monoclonal antibody at the dilution of 1:200 in PBS for 1 h at room temperature. After 1× PBS washing, the secondary antibody mixture consisting of anti-rabbit antibody conjugated with Cy3 and anti-mouse antibody conjugated with FITC at the dilution of 1:500 and 1:1000 in PBS, respectively. The coverslips were immersed in the secondary antibody for 40 min at room temperature. Following 1× PBS washing, the coverslips were mounted with Vectorshield and observed.
Statistical method
Statistical Package for the Social Sciences (SPSS) was used for analyzing the data in the study.
Results
Immunofluorescence assay of the localization of endogenous IRF-3 protein after Sendai virus infection
IRF-3 is known to be constitutively expressed in the cytoplasm in an inactive and unphosphorylated state. Upon virus infection or double-stranded RNA (dsRNA) treatment, IRF-3 is phosphorylated and translocated into the nucleus, where it activates IFN-β transcription together with other transcription factors. 10 Figure 1 shows the immunofluorescence staining of the expression of IRF-3 after Sendai virus infection. Furthermore, nucleic IRF-3 is shown in Figure 1(a) and (b), which represents the active form of IRF-3. HeLa cells on one coverslip did not show an identical protein intensity of endogenous IRF-3. Some cells strongly expressed IRF-3, while others revealed a weak expression of IRF-3. Even if both cells in Figure 1(a) and (b) were infected with the Sendai virus at 100%, which has been confirmed by experiments practiced under the same transfection and infection conditions, the nuclear translocation of IRF-3 in Figure 1(b) will distinctly be more than that in Figure 1(a). Hence, it appears impossible to induce all HeLa cells to express nuclear translocated IRF-3 after Sendai virus infection, which is regarded as a signal for the activation of IRF-3. Hence, Sendai virus infection may be a critical stimulus for the activation of IRF-3. However, the activation of IRF-3 could not be achieved by the Sendai virus infection alone. Other agents that have roles in the activation of IRF-3 work together with dsRNA virus infection.

Immunofluorescence of Hela cells transfected with or without the PSG5 plasmid and infected with Sendai virus for different hours (n = 20). Subcellular localization of IRF-3 monitored by fluorescence microscopy at ×40 objective: (a) at 18 h after Sendai virus infection, which represents the highest nucleic IRF-3 rate in the HeLa cells with viral infection alone; and (b) at 24 h after Sendai virus infection, which represents the highest nucleic IRF-3 rate in the HeLa cells transfected with the pSG5 vector (MOI = 5.0).
The cooperation of transfection and virus infection greatly enhances the nuclear translocation of IRF-3
The activation of IRF-3 after Sendai virus infection is a time-dependent course. 11 In order to obtain the highest nuclear translocation rate of IRF-3, two groups of cells were prepared. HeLa cells without transfection were assigned as group A, and HeLa cells transfected with the pSG5 vector were assigned as group B. A time-course experiment was performed to determine the optimum time to observe the nuclear translocation of IRF-3 after virus infection. According to the results shown in Figure 2, the highest nuclear translocation rate of IRF-3 in HeLa cells without transfection (Figure 2(a)) was approximately 17%, which appeared at 18 h after Sendai virus infection, while the rate was 67% in HeLa cells transfected with the PSG5 vector (Figure 2(b)), which appeared at 24 h post–Sendai virus infection. A significant difference was found between groups A and B (P < 0.001), and consistent results were obtained with different MOIs (1.25, 2.5, and 5.0). In the HeLa cells transfected with the pSG5 vector, but without Sendai virus infection, the observed nuclear IRF-3 was only approximately 3%. This suggests that IRF-3 activation was caused by the transfection process, in addition to the Sendai virus infection.

Immunofluorescence of Hela cells infected with the Sendai virus showing the time dependence of the nuclear translocation of IRF-3 (n = 20): (a) HeLa cells and (b) Hela cells transfected with the plasmid PSG5 vector. Cells were fixed at different time points at post–Sendai virus infection (4, 8, 12, 18, 24, and 36 h). The three different pattern bars represent different MOIs (1.25, 2.5, and 5.0). IRF-3 revealed different nuclear translocation rates at different time points. Twenty experiments with the mean CV (15%).
NS3/4A expression in HeLa cells could suppress the nuclear translocation of endogenous IRF-3
Figure 3 shows the double staining result of the immunofluorescence assay (IFA). The red Cy3 signal in Figure 3(a) represents the expression of endogenous IRF-3, and the green FITC signal in Figure 3(c) represents the expression of exogenous NS3/4A. NS3/4A mainly localized in the cytoplasm, while IRF-3 localized in both the cytoplasm and nucleus. In all, 200 HeLa cells and 100 NS3/4A-expressing HeLa cells were randomly counted to obtain the nuclear translocation rates of IRF-3. HeLa cells transfected with the PSG5 vector were regarded as controls. Since the transfection of pSG5 could greatly enhance the nuclear translocation of endogenous IRF-3 (Figure 2), transfection efficiency could interfere with the final rate obtained. There was no effective way to determine whether or not a HeLa cell was transfected with pSG5. Hence, cells were randomly counted to a total number of 200, which included both transfected cells and non-transfected cells. In investigating the role of NS3/4A, the control group should also include the transfected cells alone. A transfection efficiency of 50% has always been achieved under this experimental condition. The nuclear staining of IRF-3 in HeLa cells transfected with pSG5 was 45%. Therefore, it was provisionally concluded that this procedure (plasmid transfection and Sendai virus infection) mediated IRF-3 nuclear translocation in >90% of cells. Compared with the 90% nuclear translocation rate of endogenous IRF-3 in control cells, HeLa cells expressing NS3/4A manifested a decreased nuclear IRF-3 (30%); the difference was statistically significant (P < 0.001). This result indicates that the activation of endogenous IRF-3 could be inhibited by the expression of HCV NS3/4A. Furthermore, the nucleic IRF-3 rate in cells that had different NS3/4A intensities was compared (Figure 4). Among cells with NS3/4A expression, the intensity of NS3/4A was not at the same degree. Hence, NS3/4A-expressing cells were divided into two groups, according to the intensity of the expression (weak and strong). Then, nucleic IRF-3 expressing cells were counted in the two groups with a total count of 100 cells separately. The nuclear translocation rate of IRF-3 in the strong and weak groups was 25% and 62%, respectively, and the difference was statistically significant (P < 0.05). This result also supports our conclusion that the activation of endogenous IRF-3 could be downregulated by HCV NS3/4A protease.
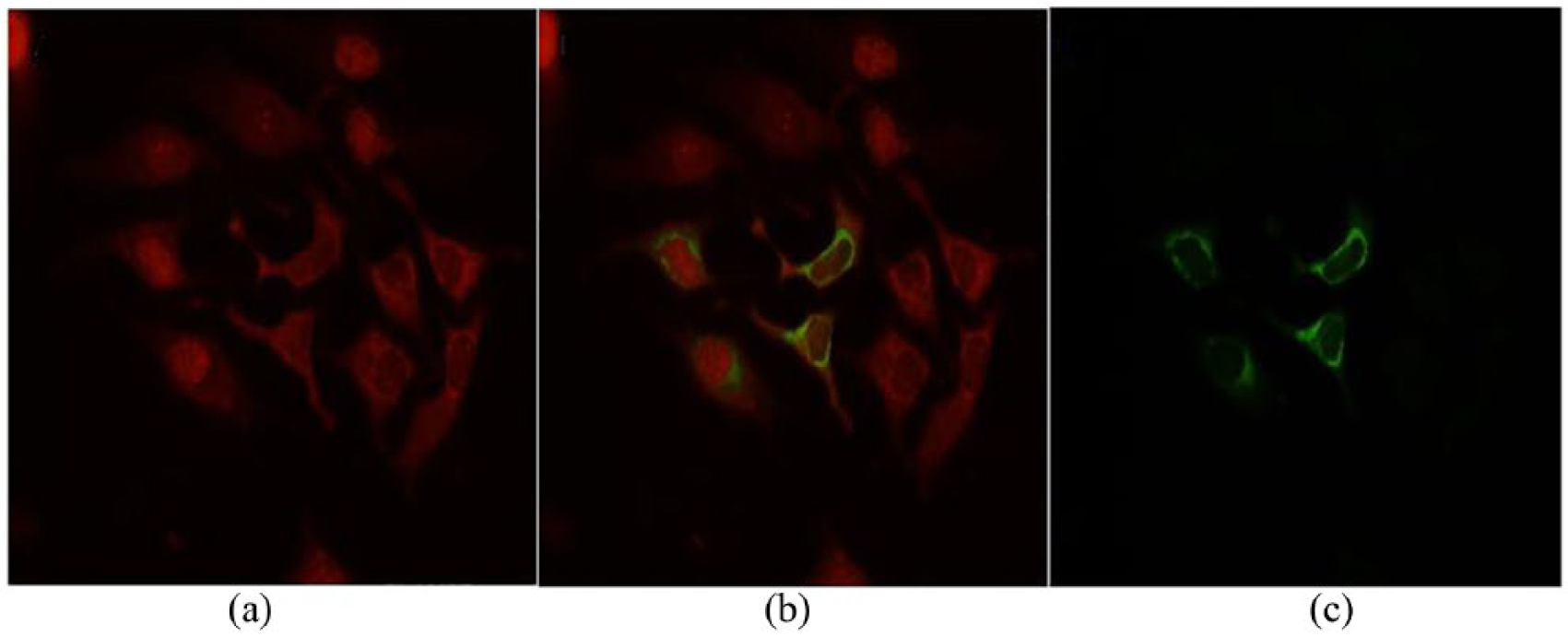
Immunofluorescence analysis showing the subcellular localization of NS3/4A and IRF-3 proteins in HeLa cells after Sendai virus infection (n = 20). (a) IRF-3 protein labeled with Cy3 in HeLa cells was observed by fluorescence microscopy at a wavelength of 550 nm. (b) NS3/4A proteins labeled with FITC in Hela cells were observed at a wavelength of 450 nm. (c) The merging of (a) and (b) shows the interaction between IRF-3 and NS3/4A. The cell-seeded coverslips were first incubated with the primary antibody mixture of rabbit anti-IRF-3 polyclonal antibody and mouse anti-NS3 monoclonal antibody at the dilution of 1:200 in PBS for 1 h at room temperature. After 1× PBS washing, cells were incubated with the secondary antibody mixture, which consists of anti-rabbit antibody conjugated with Cy3 and anti-mouse antibody conjugated with FITC at a dilution of 1:500 and 1:1000 in PBS, respectively. The coverslips were immersed in the secondary antibody for 40 min at room temperature. Following 1× PBS washing, the coverslips were mounted with Vectorshield and observed.

The immunofluorescence analysis shows the inhibition of the nuclear translocation of endogenous IRF-3 by NS3/4A protease (n = 20). Control group: HeLa cells were transfected with the pSG5 vector. All groups: HeLa cells were transfected with pSG5-NS3/4A. Weak group: HeLa cells were transfected with pSG5-NS3/4A and exhibited a relatively low expression of NS3/4A protease. Strong group: HeLa cells were transfected with pSG5-NS3/4A and exhibited a strong expression of NS3/4A protease. The nuclear translocation rate in the strong group was significantly lower than that in the weak group (P < 0.05). Twenty experiments with the mean CV (15%).
Discussion
By definition, viruses are unable to replicate on their own and must enter a host cell and use the host cell’s macromolecular machinery and energy supply to replicate. Sendai virus, a murine parainfluenza virus type 1, is a single-stranded negative sense RNA virus that has been confirmed to induce the transcription of IFN-α/β.12,13,15 In the present time-course study, as shown in Figure 1, HeLa cells that were either untransfected or transfected with the PSG5 vector exhibited time-dependent characteristics in the nuclear translocation rate of IRF-3. The highest rate was discovered at 18 to 24 h post-infection. The peak time is the direct evidence that IRF-3 actually responds to invasion of the virus. C-terminal phosphorylated IRF-3 is subject to proteasome-mediated degradation.11,14 Furthermore, IRF-3 protein exhibits dual roles in the host’s immunity response to virus invasion: the transcription induction of IFN-α/β and the apoptosis induction of cells. These two consequences are totally different: The former keeps the cell alive, and the latter destroys the cell. It is possible that during the earlier defensive period, the main function of the IRF-3 pathway is to induce the production of IFN-α/β, which causes antiviral resistance by activating cellular genes that encode antiviral proteins. Afterward, once the immune defensive system of the host has lost control of the virus replication, IRF-3 begins to induce apoptosis, which could also hold down the replication of the virus through the death of virus-infected cells.
Regarding the relationship between HCV NS3/4A protein and endogenous IRF-3, our results have shown that the expression of NS3/4A protease inhibits the activation of endogenous IRF-3, leading to the evasion of the virus from the host defense mechanism.
Therefore, HCV NS3/4A could be a novel and attractive therapeutic target, in which the inhibition of NS3/4A may not only block HCV viral replication but also restore IRF-3 antiviral function in HCV infection. 7
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.