
Abstract
Janus kinase inhibitors (JAKis), such as baricitinib, target the Janus kinase/signal transducers and activators of transcription pathway to regulate proinflammatory cytokines and T-helper cell activity, thereby reducing inflammation. Despite their therapeutic benefits, inflammatory demyelinating diseases of the central nervous system (CNS) have been reported in association with JAKi treatment, most previously involving tofacitinib in patients with rheumatoid arthritis. We report the first case of a CNS inflammatory demyelinating disease that was possibly exacerbated by baricitinib, underscoring the need for heightened clinical vigilance and close monitoring of patients receiving JAKi therapy.
Introduction
The expansion of biologic therapies for cancer and autoimmune diseases has been accompanied by a rise in iatrogenic demyelinating disorders of the central nervous system (CNS), such as multiple sclerosis (MS), neuromyelitis optica spectrum disorder, and autoimmune encephalitis. 1 Baricitinib is an orally administered, small-molecule Janus kinase inhibitor (JAKi). It exhibits greater inhibition potency against JAK1 and JAK2 subtypes than JAK3 and tyrosine kinase 2 (TYK2). By modulating key inflammatory signaling pathways, baricitinib induces remission in rheumatoid arthritis, atopic dermatitis, and systemic lupus erythematosus. Baricitinib is also approved for use in adults with severe alopecia areata and in the United States for hospitalized patients with coronavirus disease 2019 (COVID-19).2,3
While JAKis are effective in treating inflammatory and autoimmune diseases, their profound impact on cytokine signaling and immune regulation may predispose individuals to CNS inflammation in susceptible individuals, necessitating careful monitoring and management of potential neurological side effects. 4 To increase awareness and knowledge of this topic, we report the first case of CNS inflammatory demyelinating disease that manifested in the setting of baricitinib therapy for alopecia areata.
Case history
A 65-year-old male with a past medical history of atrial fibrillation, hypertension, hyperlipidemia, coronary artery disease, prediabetes, Hashimoto's disease, and neurogenic bladder began treatment with oral baricitinib for alopecia areata universalis in early 2023. The patient had previously failed topical, intralesional, and intramuscular steroids, topical tacrolimus, topical tofacitinib, and platelet-rich plasma.
Eighteen months after starting baricitinib, he experienced painless vision loss in the left eye, which worsened over the course of one week. A month prior to this event, the patient had a streptococcal pharyngitis followed by a viral upper respiratory infection. He presented to the emergency department, where neurological examination was remarkable for a relative afferent pupillary defect with decreased visual acuity in the left eye (20/200) and diminished light-touch sensation in the lower extremities. The ophthalmologic examination showed a trace of disc edema, blurred disc margins with faint borders, and mild pallor, along with atrophy of the left optic nerve.
Comprehensive blood and cerebrospinal fluid studies were unremarkable except for a mildly low vitamin D level (Table 1). Computed tomography (CT) angiogram of the head and serologic workup for vasculitis, along with chest-abdomen-pelvis CT, were unremarkable. A 3-Tesla (3T) magnetic resonance imaging (MRI) of the brain, orbits, and total spine obtained before and after the injection of the contrast agent gadolinium diethylenetriaminepentaacetic acid (Gd-DTPA) showed multiple contrast-enhancing lesions (CELs) in the brain (Figure 1(a)–(d)), cervical spinal cord (Figure 1(e) and (f)), and left optic nerve (Figure 1(g) and (h)), suggesting a nearly simultaneous occurrence.
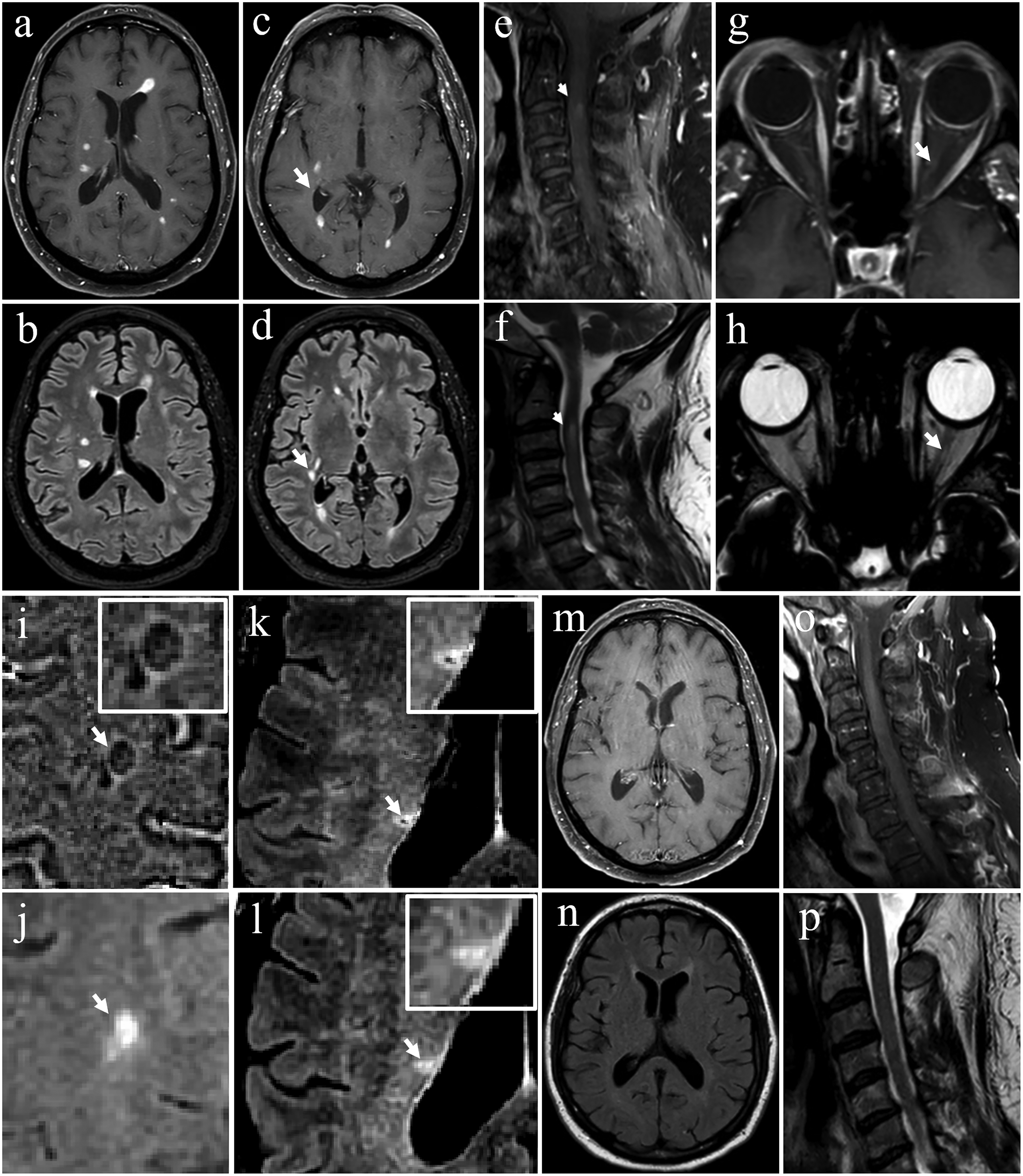
Brain (a–d), cervical spine (e, f), orbits (g, h), MRI at the time of admission, brain MRI a month later (i–l), brain (m,n), and cervical spine MRIs (o,p) six months later. (a, c) T1-w spin echo post contrast agent Gd-DTPA showing multiple CELs and (b, d) T2-w FLAIR sequence showing additional non-CELs (white arrow as an example). (e) T1-w post-Gd-DTPA and (f) T2-w sagittal cervical spine MRI showing a CEL (white arrow) at the level of C3. (g) axial T1-w post-Gd-DTPA and (h) T2-w orbit MRI showing a fading CEL (white arrow). (i) axial phase shows a PRL (white inset and arrow) and (j) axial T2-w FLAIR shows the corresponding PRL; (k) axial susceptibility weighted imaging multiplied by a T2-w FLAIR is also shown in (l) displaying an exemplary lesion with a central vein sign (white inset and arrow). (m) Axial brain T1-w post-Gd-DTPA, (n) brain T2-w FLAIR, (o) sagittal cervical spine T1-w post-Gd-DTPA (p) T2-w images. Post-Gd-DTPA scans show near-complete CELs resolution six months later.
Serological and cerebrospinal fluid test results.

ANA: antinuclear antibody; ANCA: anti-neutrophil cytoplasmic antibody; RF: rheumatoid factor; CSF: cerebrospinal fluid; VDRL: Venereal Disease Research Laboratory.
*Fluorescence-activated cell sorting (FACS) assay by Mayo Clinic.
**CSF and serum paraneoplastic panel were performed at Mayo Clinic Laboratories, Rochester, Minnesota. https://www.mayocliniclabs.com/test-catalog/overview/37430.
#Below normal value. ##Above normal value.
Specifically, we counted 25 T2 lesions, 20 of which corresponded to CELs. Of the five non-CELs, two were typical for MS, located in the deep white matter of the right temporal lobe and the right periventricular area (frontal horn). The remaining three lesions were small and non-MS-specific (deep corona radiata).
Baricitinib was discontinued, and the patient received intravenous methylprednisolone (1 g daily for six days), followed by a 46-day oral prednisone taper. A week after symptom onset, left-eye visual acuity improved to 20/25, and fundoscopic examination showed optic nerve pallor without disc edema. Using optical coherence tomography, average retinal nerve fiber layer (RNFL) thicknesses of 87 and 64 µm were measured in the right and left eyes, respectively. The peripapillary RNFL was thin in the superior and inferior quadrants, and the left optic nerve was atrophied. These findings have remained stable to the time of this report. The patient's neurological examination returned to normal, and a follow-up brain MRI one month later showed resolution of all CELs, with most of them not being followed by a T2 abnormality (data not shown). Phase maps showed a paramagnetic rim lesion (PRL; Figure 1(i) and (j)), while the susceptibility-weighted image was positive for the central vein sign (CVS) based on the Select-3 criteria (Figure 1(k) and (l)).
Five months later, the patient presented with complaints of progressive stiffness and weakness of the lower extremities, along with diffuse, stabbing joint pain, most notably in the left shoulder, left upper back, hips, and knees. Symptoms had begun three months earlier following a streptococcal throat infection and had worsened significantly after a subsequent influenza infection a few weeks before presentation.
Blood work was remarkable for an elevated erythrocyte sedimentation rate (ESR) up to 58 mm/h (normal: 1–33 mm/h) and C-reactive protein (CRP) up to 51.9 mg/L (normal: 0–5 mg/L). A diagnosis of polymyalgia rheumatica was made based on clinical and serologic data, and low-dose prednisone (15 mg daily) was initiated. Rapid symptomatic improvement and gradual normalization of ESR and CRP values were observed within 16 days of prednisone initiation.
A new MRI of the brain (Figure 1(m) and (n)), cervical spine (Figure 1(o) and (p)), and thoracic spine with and without Gd-DTPA showed further improvement of prior lesions.
Adverse drug reaction probability scale (Naranjo scale) assessment
To increase accuracy regarding causality, we applied the adverse drug reaction probability scale (Naranjo scale) to this case. The total score was 4, corresponding to a possible adverse drug reaction. 5 We counted a prior report in the JAK inhibitor class as (+1), the temporal relationship with onset 18 months after starting baricitinib as (+2), clinical and radiological improvement after discontinuing the drug and administering intravenous methylprednisolone as (+1), and objective MRI confirmation as (+1). We reasoned that the presence of paraclinical evidence of MS, such as PRLs, neurogenic bladder, and optic nerve atrophy, contributed as (−1), as these findings suggested the possibility of alternative causes. A near-complete resolution of enhancement within one month after baricitinib discontinuation supports a drug-related process while acknowledging competing explanations. The relevant timing and imaging are documented in the case history and in Figure 1.
Discussion
Our case highlights an instance of CNS inflammatory demyelination in the setting of baricitinib use. Although causation cannot be definitively established, the temporal association between JAKi initiation and the onset of neurological symptoms raises concern for a “possible” association. 5 Our case is unique in that it presents features of both chronic and acute-on-chronic disease, prompting the question of whether this represented a pre-existing, subclinical demyelinating process that acutely reactivated during treatment with baricitinib, or a completely new-onset disease.
The patient's age, painless vision loss, high number of CELs with subsequent near-complete resolution of abnormalities on T2-weighted images, and absence of oligoclonal bands argue against MS and suggest a potentially iatrogenic origin. The clinical presentation also does not support a diagnosis of acute disseminated encephalomyelitis. 6
It is noteworthy, however, that the patient had a history of neurogenic bladder (typically seen in MS), presented with atrophy of the left optic nerve (suggestive of a prior subclinical optic neuritis), and had a brain MRI positive for PRL and CVS (chronic findings specific for MS). These elements together indicate that the patient may have harbored a latent, relatively quiescent demyelinating condition that became clinically overt following exposure to baricitinib.
Notably, the optic neuritis presented a challenging clinical dilemma due to its painless nature. Although he had several risk factors for an ischemic etiology of his vision loss, the absence of disc edema at the time of the acute episode and the drastic, steroid-responsive visual improvement argued against it. All these findings suggest the possibility of a pre-existing silent demyelinating condition that was unmasked during baricitinib therapy. This would also not be surprising given the high tendency of our patient to suffer from autoimmune diseases, for example, Hashimoto's thyroiditis, alopecia areata, and polymyalgia rheumatica.
Interestingly, both the CNS demyelinating episode and subsequent polymyalgia rheumatica developed shortly after a streptococcal and viral upper respiratory infection. While this temporal relationship may suggest a contributing role for postinfectious immune activation, the patient's prolonged exposure to baricitinib likely facilitated immune disequilibrium, allowing these events to manifest more easily.
Prior reports of JAKi-associated demyelination are limited but informative. Massoud et al. 7 described the first case of CNS demyelination associated with tofacitinib, in which a monophasic clinical course, absence of oligoclonal bands, and complete recovery after drug discontinuation and steroids use favored an iatrogenic etiology rather than MS. Using the Naranjo adverse drug reaction probability scale, that case was scored as “probable.” Similar to our case, the latency between drug initiation and neurological symptoms was prolonged, highlighting that iatrogenic demyelination can emerge even years after exposure.
More recently, a pharmacovigilance analysis using the global VigiBase database identified memory impairment and peripheral neuropathy in association with tofacitinib and ruxolitinib. On the contrary, baricitinib showed relatively few neurological adverse signals, such as sciatica and post-herpetic neuralgia. 8 In pharmacovigilance, a signal refers to a statistical alert generated when a particular adverse event is reported disproportionately often for a given drug compared with all other drugs and thus indicates a potential drug event association requiring further study rather than confirmed causation. 8 Although causality could not be definitively proven in that study, these findings highlight that neurological complications, including demyelination, are biologically plausible and may differ across JAKis. Together, these prior reports reinforce the need for vigilance in monitoring neurological events during JAKi therapy. 8
Consideration of the safety and appropriateness of use of JAKis in the setting of demyelinating disease is further complicated by the notion that JAKis may possess neuroprotective properties and facilitate remyelination, 9 with contradictory reports indicating their involvement in demyelinating processes. 7 In a cuprizone-induced model of MS, tofacitinib was shown to enhance remyelination, improve motor function, and reduce inflammatory cytokine signaling through suppression of signal transducers and activators of transcription (STAT)-3 and STAT-5 phosphorylation. 9 In this study, tofacitinib rescued myelin basic protein loss, ameliorated motor impairment, and lowered matrix metalloproteinase activity without evidence of systemic toxicity, supporting the hypothesis that JAK/STAT inhibition may confer neuroprotection. 9
The JAK family includes JAK1, JAK2, JAK3, and TYK2, which mediate intracellular signal transduction of numerous cytokines. 10 All cytokine receptors, regardless of type, are linked to one or more JAKs to enable signal transduction. Upon activation and transphosphorylation, JAKs initiate the recruitment of STATs, leading to their dimerization, nuclear translocation, and the initiation of transcriptional responses. 11 JAK signaling is meticulously regulated at multiple levels, and there is significant cross-talk with other intracellular pathways, the complexities of which are still being unraveled. 11 JAKis target the JAK–STAT signaling pathway, which is essential for cytokine and growth factor-mediated immune function, inflammation regulation, and cellular communication. By blocking the phosphorylation and activation of STATs, JAKis effectively disrupt inflammatory responses. However, this interference theoretically can inadvertently contribute to CNS demyelinating diseases through several mechanisms. First, JAKis can impair the signaling of protective cytokines such as type I and II interferons and interleukin-6, which are crucial for maintaining immune surveillance within the CNS, controlling autoimmunity, and combating latent viral infections. 12 Second, by inhibiting JAK1 and JAK3, JAKis weaken the immune system's ability to detect and eliminate autoreactive immune cells or latent pathogens. Third, JAKis create an imbalance in cytokine signaling by disrupting the pro/anti-inflammatory cytokine equilibrium, increasing susceptibility to demyelinating diseases. Last, despite efforts to develop selective JAKis, many still exhibit off-target effects by inhibiting additional JAK isoforms such as TYK2, leading to broader disruptions in immune and CNS signaling pathways. 4
Conclusions
Although individuals with an autoimmune disease may have a general predisposition to other immune-mediated disorders, 7 clinicians should be aware of the possibility of new-onset atypical and rare CNS demyelinating diseases as well as exacerbation of potentially pre-existing ones in the setting of JAKis use. Given the complex and contradictory nature of JAKis effects on the CNS, further research is essential to elucidate the mechanisms underlying these adverse events and to develop comprehensive safety profiles. Enhanced investigative efforts will help identify risk factors, optimize treatment protocols, and ensure the safe use of JAKis in managing autoimmune and inflammatory disorders. Our case therefore underscores the need for careful patient monitoring and further research to reconcile these opposing observations.
Footnotes
Acknowledgements
We thank our patient for consenting to this anonymous report, which will benefit individuals with CNS demyelinating disease and advance the knowledge of the medical and scientific communities.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Rozita Khalili was a clinical fellow supported by the National Multiple Sclerosis Society (RG- 1901-33190) at the time of this manuscript submission. Ahmad A. Toubasi receives unrelated funds from the Veterans Health Administration (I01CX002160-01A1). Francesca Bagnato receives unrelated funds from the National Multiple Sclerosis Society (RG-1901-33190 and RG-2407-43760), the Veterans Health Administration (I01CX002160-01A1), the Department of Defense (MS220136), and the Voros Innovation Impact Funds.