
Abstract
Background
SLIPPERS (Supratentorial Lymphocytic Inflammation with Parenchymal Perivascular Enhancement Responsive to Steroids) is a rare variant of a syndrome called CLIPPERS (Chronic Lymphocytic Inflammation with Ponsine Perivascular Enhancement Responsive to Steroids). SLIPPERS is characterized by distinct supratentorial lesions that share radiological and pathological characteristics with CLIPPERS. The ongoing issue is whether these syndromes should be considered as a distinct disease entity or simply a form for a variety of underlying conditions such as granulomatosis, vasculitis, and infectious diseases.
Case
We present a unique case of SLIPPERS observed in a 26-year-old woman with no notable medical or familial background. Laboratory findings ruled out certain diseases from the list of differentials and cranial MRI showed T2 hyperintense areas with linear-patchy enhancements, a pattern consistent with SLIPPERS syndrome. Consequently, patient was diagnosed with SLIPPERS syndrome and received methylprednisolone therapy.
Conclusion
Both SLIPPERS and CLIPPERS are complicated syndromes posing diagnostic challenges and requiring careful investigation to avoid misdiagnosis. Following a thorough differential diagnosis, appropriate treatment can be initiated, and follow-up is required.
Introduction
Within the central nervous system diseases, there exists an uncommon phenomenon recognized as Supratentorial Lymphocytic Inflammation with Parenchymal Perivascular Enhancement Responsive to Steroids (SLIPPERS) syndrome. 1 This condition, which remains inadequately comprehended, is situated within the realm of neuroinflammatory disorders, posing a distinctive diagnostic dilemma for neurologists.
While sharing similarities regarding to their immunopathological mechanisms, such as CD3+, CD4+ T lymphocytes, CD20+ B lymphocytes infiltration, and gadolinium-enhancing lesions, these syndromes exhibit distinct anatomical localizations, implying that SLIPPERS may follow a distinct pathway of its own.2,3 SLIPPERS’ defining characteristic lies in its selective predilection for the supratentorial brain regions, encompassing the cerebral hemispheres and sparing the brainstem and cerebellum. 4 This distinct topographical distribution throws complexity over diagnosis, often mimicking other neurological and inflammatory conditions such as CNS vasculitis, anti-glial fibrillary acidic protein (GFAP)-associated disease, vascular anomalies, and granulomatous diseases. 5
The complexity of SLIPPERS syndrome is not fully understood, as its unique clinical characteristics, neuroimaging features, and underlying microscopic aspects distinguish it from other conditions. Various clinical symptoms can be observed, including focal neurological deficits, cognitive decline, and seizures. 4 Magnetic resonance imaging (MRI) stands as the cornerstone, revealing a characteristic pattern of multiple, punctate, gadolinium-enhancing lesions scattered throughout the supratentorial white matter and gray matter, often described as a “peppering” pattern. 6
Brain biopsy may be required due to the difficulty of diagnosis and challenging in differential diagnosis with other diseases. However, this option may pose inconvenience for the patient, potentially leading to refusal. A detailed MRI evaluation, along with close follow-up after steroid treatment, allows for a definite diagnosis without the need for a cerebral biopsy, as in the case of our patient. On the other hand, the biopsy may reveal perivascular inflammation with the presence of CD4+/CD8+ T lymphocytes with CD68 + and infiltrative CD3+ T lymphocytes, accompanied by reactive astrocytes and minimal to absent granulocytes, potentially important for the definitive diagnosis.1,7 In the treatment of the SLIPPERS, corticosteroids have a promising role in inducing remission and managing relapses. 7
We aimed to raise physician's knowledge by emphasizing the difficulty of conducting a deep differential diagnosis for SLIPPERS and the significance of long-term follow-up based on this case.
Case presentation
A 26-year-old woman presented to the neurology outpatient clinic in September 2021 with the complaint of headache. Her medical history includes urticaria, residual neck pain from a prior traffic accident, and post-vaccination panic attacks, which are unrelated for the diagnosis. In the family history, there was no remarkable disease. She was alert and cooperative. Her visual acuity and cranial nerve examination revealed no abnormalities. In the assessment of the pyramidal system, muscle strength was found to be normal, with increased deep tendon reflexes noted bilaterally. The Hoffman test yielded bilateral positive, and the plantar reflex was bilaterally unresponsive, with a 1–2 beat Aschilles clonus present in the right lower extremity.
A cranial MRI performed in November 2021, revealed two discrete, heterogeneously enhancing lesions in the left frontal and parietal lobes (Figure 1). This atypical presentation posed a diagnostic dilemma, raising suspicion for demyelinating, infectious, granulomatosus diseases, vasculitis or developmental vascular anomalies.

Cranial MRI in September 2021 demonstrating heterogeneously enhanced lesions in the left frontal and parietal lobes.
Laboratory investigations yielded positive Anti-SSA antibodies, suggesting a potential autoimmune etiology. The patient did not have dry mouth or dry eyes suggestive of Sjögren's syndrome, and was also thoroughly evaluated by a rheumatologist, who ruled out Sjögren's syndrome. The absence of clinical signs, negative inflammatory markers, the lack of specific autoantibodies like NMO and MOG-Ig G and normal cervical and thoracic MRI emphasized to exclude differential diagnoses such as vasculitis, infectious diseases, neuromyelitis optica spectrum disorder (NMOSD) and MOG-associated diseases (MOGAD). By the way the MOG antibody was analyzed using the live cell assay method. The aquaporin 4 (AQP4) antibody was evaluated by the cell-based immunofluorescence assay twice. A lumbar puncture was recommended to the patient, but the patient did not consent to the procedure. Given the discrepancy between clinical and imaging findings, the neuroradiology council recommended close clinical and radiological follow-up.
Subsequent MRIs performed over the following months, including April and November 2022, demonstrated persistent lesions, prompting consideration of diagnoses such as autoimmune encephalitis, vascular anomalies, tumors or even perivenular sarcoidosis. Onconeural antibodies and antibodies against neuropil were negative. Additionally, computerized tomography (CT) scans of the chest and abdomen revealed no abnormalities, with no evidence of lymphadenopathy or findings suggestive of lymphoma, a primary tumor, or systemic sarcoidosis. The lack of clinical progression and specific supporting evidence for these diseases made the possibilities less certain. In MR Spectroscopy, no significant features were detected. In digital cerebral angiography, no significant vascular pathology was detected and possible vascular anomalies have been ruled out.
In November 2023, a comprehensive comparison of serial MRIs revealed not only lesion stability but also subtle T2 hyperintense areas with linear-patchy enhancements in the left cerebral hemisphere (Figure 2). This specific pattern, known as “peppers pattern” in the literature, combined with the persistent headache and subtle neurological findings, strongly suggested the diagnosis of SLIPPERS syndrome. The presence of stable, non-enhancing T2 hyperintensities alongside patchy contrast enhancement patterns is increasingly recognized as a characteristic feature of SLIPPERS syndrome, aiding in its differentiation from other entities with similar imaging findings. In light of the prevailing therapeutic approach for this syndrome, which prioritizes the administration of steroids as the primary first-line intervention, we initiated steroid therapy. This decision was guided by the necessity to ascertain a definitive diagnosis, achieved through observing the clinical response of the lesions to the steroid treatment. Although a definitive diagnosis could have been established with a brain biopsy, the patient declined to undergo the procedure for diagnostic purposes. Since the biopsy required for a definitive diagnosis of the disease was not performed, it was considered probable SLIPPERS, and the treatment was planned accordingly.

In November 2023, cranial MRI showing stable T2 hyperintense areas with linear-patchy enhancements, consistent with SLIPPERS syndrome (peppering pattern).
Initially, 1 gram intravenous methylprednisolone (IVMP) per day was administered for 5 days, followed by 1 gram IVMP per month for 3 doses. Although there is no clear information in the literature regarding the optimal treatment duration, the treatment was continued with 1 pulse of steroid per month for 3 months.1,4,6 In our case, after 5 days of pulse steroid therapy, treatment was continued with monthly pulse therapies. Due to the patient's reluctance towards oral treatment but the necessity for continued therapy, it was mutually decided to proceed with monthly pulse treatments. Post-treatment control imaging showed that the lesion in the posterior had completely regressed. In the lesion in the frontal region, a decrease in edema, reduction in lesion size, and regression in contrast enhancement were observed (Figure 3). Upon observing a positive clinical and imaging response in the assessments conducted in the third month of treatment, a consensus was reached to recommend immunosuppressive therapy to the patient.
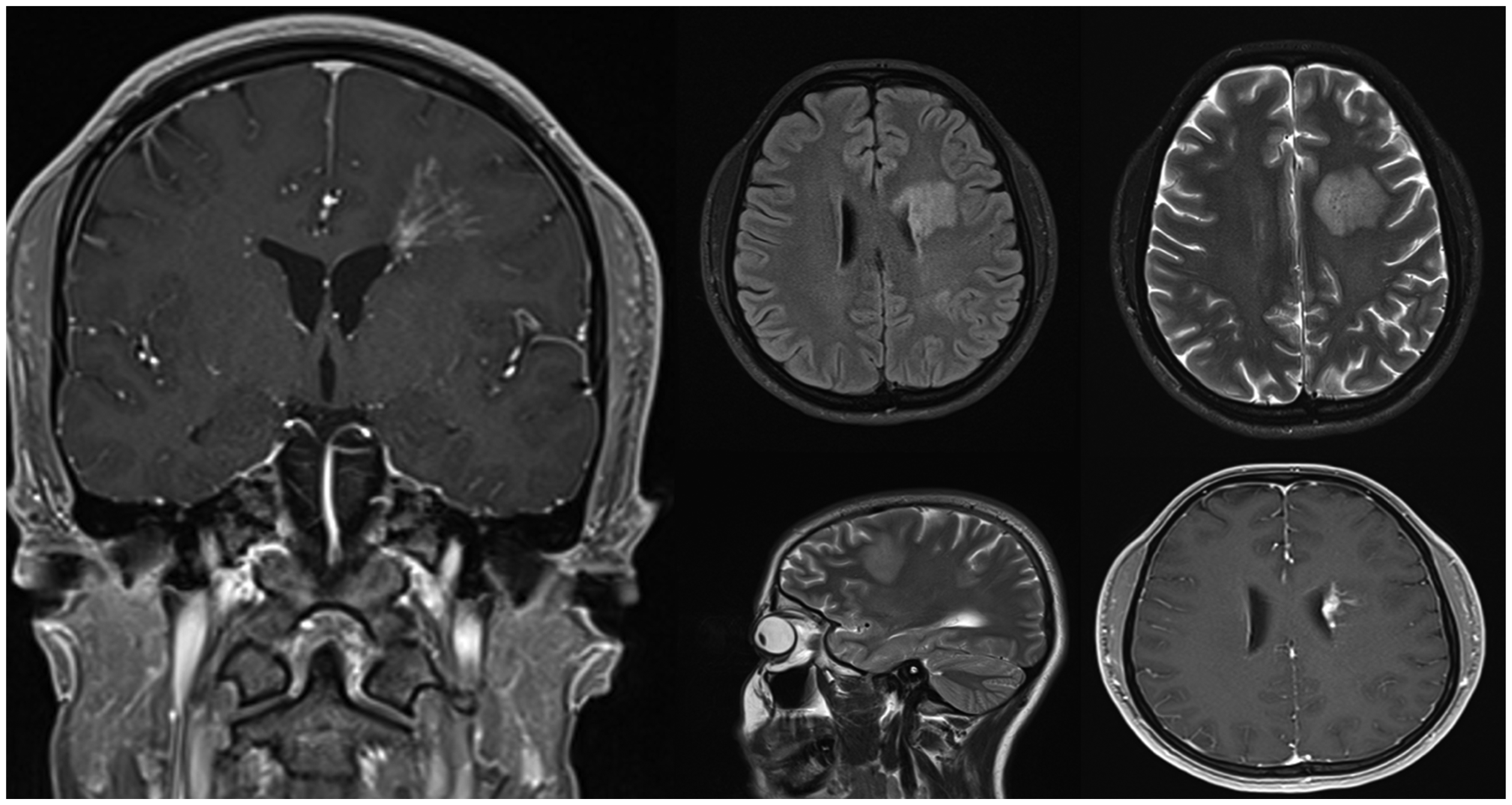
Post-treatment control MRI showing lesion in the posterior had completely regressed and in the frontal lesion regression was observed.
Discussion and conclusion
CLIPPERS represents an infrequent neurological disorder primarily affecting the pons, characterized by a unique perivascular enhancement pattern evident on MRI. While the initial characterization of CLIPPERS underscored its association with pontine involvement, subsequent observations indicate that over fifty percent of CLIPPERS cases exhibit supratentorial participation, displaying imaging patterns similar to those observed in the brainstem.2,8,9 Armand et al. (2015) published a case report detailing a patient with a CLIPPERS-like syndrome but presenting supratentorial lesions. 1 A recently published article also mentions spinal cord involvement. In terms of the area of involvement, this suggests that the CLIPPERS syndrome is not yet clearly understood. 10 Therefore, it is open to discussion whether these existing entities are manifestations of CLIPPERS syndrome in different localizations or new definitions (such as CLIPPERS with spinal involvement or CLIPPERS with hemispheric involvement). Thus, from one perspective, SLIPPERS syndrome could currently be defined as CLIPPERS with hemispheric involvement. To date, a few cases of SLIPPERS have been documented in the literature. Our report introduces the ninth patient to present new radiological and clinical findings. We believe that it is necessary to discuss whether it indicates a syndrome pointing towards demyelinating, granulomatous, tumoral, or infectious diseases, or it is a standalone disease that requires separate treatment
As SLIPPERS is a rather new rare variant of CLIPPERS, definitive diagnostic criteria have yet to be established. Thus, it would be logical to refer to the diagnostic criteria for CLIPPERS, which encompass clinical, radiological and pathological parameters. In accordance with these criteria, CLIPPERS patients typically exhibit subacute pontocerebellar lesions that display responsiveness to steroid therapy, while excluding other potential differential diagnoses. These lesions manifest as small, punctuate gadolinium enhancing nodules smaller than 3 mm in diameter, devoid of ring enhancement. If possible, a brain biopsy should be investigated to ascertain perivascular CD4+ T cell dominant inflammation present in both white and gray matter, thereby ruling out alternative explanations. We can adopt these criteria for SLIPPERS as well with the exception of the location of lesions. 11 In our case, the absence of progression observed in the differential diagnoses during the follow-up, alongside the contrast enhancement pattern, provided significant clues. Performing a thorough differential diagnosis with the support of cerebral angiography and MR spectroscopy, along with a positive response to steroid therapy, strengthened the likelihood of our diagnosis. In GFAP encephalopathy, the clinical presentation often manifest as encephalitis, meningoencephalitis, and myelitis, with the most specific manifestation being periventricular “vascular radiolucency”. 12 Through clinical and MRI assessments, GFAP encephalopathy was ruled out from differential diagnosis in our patient. We agree that testing for GFAP antibodies is necessary to exclude GFAP astrocytopathy in the differential diagnosis. However, the blood NFL/GFAP panel is not yet available in our country. This limitation presents an additional challenge in differentiating SLIPPERS diagnosis, particularly for countries where testing for these antibodies is not yet accessible.
While definitive treatment protocols have not been proposed yet for both syndromes, initial pulse steroid therapy is universally administered to induce remission following relapse, typically for 7–10 days at a daily dosage of 1 g. Maintenance therapy plays a crucial role in preventing further relapses. Kamışlı et al. adopted a regimen consistent with previous literature, transitioning to oral methylprednisolone at 1 mg/kg daily following initial therapy. Additionally, they prescribed azathioprine (100 mg/day) as a corticosteroid-sparing agent alongside a maintenance dose of methylprednisolone at 20 mg/day. While azathioprine alone may sustain remission, prolonged corticosteroid therapy appears necessary for most CLIPPERS and SLIPPERS patients, as discontinuation or tapering may precipitate disease relapse. However, the optimal duration of treatment remains unclear. 13 In the long term, the potential role of disease-modifying therapies (DMT) is under discussion. Notably, a successful case report by Didier et al. employed oral leflunomide at a weekly dosage of 100 mg following initial corticosteroid treatment, achieving clinical remission for one year. However, the lack of long-term data due to the patient's discontinuation of treatment precludes conclusive insights into its efficacy over extended periods. 14
Typically, CLIPPERS syndrome presents as a distinct entity; however, some hypotheses suggest that these syndromes may represent initial manifestations of other diseases such as autoimmune gliopathies, vasculitis, or lymphoma. 15 Currently, both CLIPPERS and SLIPPERS are regarded as syndromes, as the available evidence is not robust enough to classify them as distinct diseases. Nevertheless, as demonstrated in the literature, these syndromes may not always manifest as discrete entities. For instance, Symmonds et al. illustrated that CLIPPERS may coexist with MOGAD, altering both clinical and radiological findings. While cervical cord inflammation is commonly described in CLIPPERS, the observation of longitudinally extensive thoracolumbar cord lesions was seen in the codiagnosis of MOGAD and CLIPPERS. Later finding emphasized the necessity of MOG antibody testing in the differential diagnosis. 16 Another study by Martin et al. indicated that CLIPPERS may also coincide with NMDAR antibody encephalitis. While cerebellar ataxia is a rare symptom of NMDAR-IgG encephalitis, it is the predominant manifestation of CLIPPERS, which could be important to consider in the differential diagnosis. The emergence of overlapping autoimmune central nervous system (CNS) disorders underscores the increasing recognition of these complexities. 17 Consequently, accurate differential diagnosis becomes paramount in guiding appropriate treatment. Although both CLIPPERS and SLIPPERS are readily treatable conditions with steroids, diagnosis should be meticulously approached, considering all the aforementioned criteria and excluding other potential diseases.
The diagnosis duration of rare diseases such as SLIPPERS is prolonged. For some diseases like sarcoidosis or seronegative autoimmune diseases, the diagnosis process may take up to 10 years.18,19 In our patient, the time from the initial clinical presentation to diagnosis exceeded 2 years. Although this is quite a lengthy period, the fact that the patient was diagnosed with a rare disease like SLIPPERS makes this duration more acceptable. Rare diseases require this type of long-term monitoring, and although it is challenging for both the patient and the physician, extended follow-up is very valuable.
The unavailability of GFAP antibody testing in our country and the patient's lack of consent for lumbar puncture represent major limitations in the diagnostic process. It can also be said that these limitations may contribute to the prolonged diagnostic timeline. Additionally, the inability to perform a brain biopsy due to the patient's refusal represents another limitation in establishing a definitive differential diagnosis such as lymphoma.
In conclusion, our study provides a comprehensive analysis of probable SLIPPERS syndrome, shedding light on its intricate relationship with CLIPPERS and the challenges in diagnosis and treatment. Our findings contribute to the growing body of knowledge, emphasizing the importance of accurate differential diagnosis for optimal patient care.
Footnotes
Acknowledge
We extend our heartfelt gratitude to
Author contributions
Drafting, writing: İA, AA, Supervision: AA, Data: AS, MSA, Literature Searching: AS, İA
Data availability
Data will be provided by request via mail to corresponding author.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.