
Abstract
Background
HTLV1-associated myelitis (HAM) is a slowly progressive myelopathy in which spinal cord MRI demonstrates no lesion or atrophy.
Objective
We examined the overlap between NMOSD features and HTLV1 infection.
Methods
We included all HTLV1-infected patients recruited in French West Indies (FWI) or referred from different centers, and suffering from at least one NMOSD feature. Literature connecting HTLV1-infection and NMOSD was reviewed.
Results
We included six NMOSD-like HAM with acute onset, seronegative against AQP4 and MOG-Abs. All displayed extensive longitudinal myelitis, and the optic nerve was involved in three. We gathered 39 cases of NMOSD-like HAM patients from the literature. Atypical signs of HAM were relapses (15.4%), sensory level (50%), upper limb symptoms (35.9%), optic neuritis (10.2%). Typical lesions involved lateral funiculi and featured a double rope sign (56.3%).
Conclusion
We propose that acute onset of NMOSD-like HAM could be more frequent than expected and should be evoked in high-risk patients. Extensive but often transient cord lesions could be the hallmark of an excessive inflammation of the funiculi targeted by HTLV1 infection. Although usually minor, a few HAM cases demonstrate specific MRI lesions, and the most severe cases may mimic NMOSD attacks.
Keywords
Introduction
HTLV1-associated myelitis (HAM) is a slowly progressive myelopathy characterized by a clinical triad associating lower limb weakness and spasticity, bowel and bladder dysfunction, and minimal distal sensory dysfunction in the lower limbs without sensory level. Spinal cord (SC) MRI is usually normal and cord swelling is an exclusion criterion. 1 Although progression of HAM is not uniform, an acute course is a highly uncommon feature of HAM, in contrast with neuromyelitis optica spectrum disorders (NMOSD).
We report for the first time a single-center series of seronegative NMOSD-like attacks in HTLV1-infected patients. Thanks to the first complete literature review of this topic, we have identified a new MRI feature: longitudinal extensive HTLV1-associated myelitis (LEHAM). We propose that the spectrum of HAM should be broadened into a continuum of presentations extending to NMOSD-like HAM onset with LEHAM.
Methods
We included all NMOSD patients (Wingerchuk criteria, 2015 2 ) infected by HTLV1 and followed up in Centre Hospitalier Universitaire de Martinique, Fort-de-France, French West Indies (FWI). Proviral load in CSF was assessed as previously described. 3 Assessment of antibodies against AQP4 and MOG was obtained from frozen serum collected prospectively (Patient 1–5) and sent to Mayo Clinic, Rochester, for quantitation. 4 One patient was referred from Metropolitan France (Patient 6), and AQP4 and MOG antibodies were assessed by cell-based assay at the INSERM U842 laboratory in Lyon, France. MRI scans were acquired in different centers, and multiple upgrades were done between 2005 and 2021, so sequence parameters time were for routine clinical use but were not standardized for the purpose of the study. All MRI scans were performed on a 1.5 Tesla magnet or higher, and included sagittal T1-weighted (w), T2w and post-contrast T1w images. Study parameters were kept simple and unambiguous so that they were less dependent on the availability of MRI machines. All patients provided written informed consent.
A PubMed search was performed to identify cases of HTLV1-infected patients sharing features with NMOSD. Methods are given in Supplementary Data S1. Finally, 41 non-duplicate HTLV1-infected cases were gathered (Supplementary Table S2). We used a normal approximation of the binomial calculation to estimate the confidence interval for a proportion (95% confidence level).
Results
Patients
Six patients were included (Table 1; full data are provided in Supplementary Data S3): five were Afro-Caribbean patients, all of them born and living in the FWI. One was Caucasian living in Metropolitan France. All patients were positive against HTLV1 in serum, but negative against MOG and AQP4-Abs. Atypical signs for HAM were: acute onset (≤1 month): 4; sensory level: 6; upper limbs involvement: 2; optic neuritis: 3. Spinal MRI demonstrated longitudinally extended (≥3 segments) myelitis in all patients (Figures 1 and 2). Other MRI signs were: cervical involvement: 3; edema: 4; contrast-uptake: 4; double rope sign: 2.
Main characteristics of NMOSD-like HAM patients.

Cs: corticosteroids; BS: Brown-Séquard syndrome; Mitox: mitoxantrone; ON: optic neuritis; PLEX: plasma exchange; RTX: rituximab; sph: sphincter impairment.
aAge was rounded to the closest decade for the purpose of anonymization.
bOutcome was EDSS (when available), progression or death; improvement was subjectively stratified to null, mild, moderate or major.

Brain and spinal cord MRI. Patient 2. (a and b) Extensive dorsal pencil-thin myelitis (T7 to L1) with edema, cloud-like contrast-uptake, and anterior predominance, suggesting involvement of pyramidal tracts. Patient 3. (c and e) Relapse involving optic chiasma, (d to f) medullary pyramids, and (f) cervical cord with clear predominance to pyramidal tracts, featuring the double ropes sign (arrows). (g and h) Cervical and dorsal longitudinal myelitis extending from medulla to T11, with diffuse edema and cloud-like contrast uptake. (g inset) Lesion of right corticospinal tract at thoracic level. (i) Two-year follow-up demonstrated almost complete regression of lesions and diffuse cord atrophy. Patient 4. (j) Extensive cervical myelitis (C2 to T1) with cord swelling. Absence of contrast-enhancement (not shown). MRI sequences were: FLAIR (c to f), T2-weighted (a, g and inset, i-j) and contrast-enhanced T1-weighted (b and h).

Brain and spinal cord MRI. Patient 6. (a) Longitudinal extensive myelitis (medulla to conus) with major edema. (b and c) Ill-defined contrast-uptake extending along the central cord and anterior medulla. (d to e) Systematization to pyramidal tracts is best observed on axial slices at medulla and C1 levels (double ropes sign, arrows). (f) Follow-up obtained at day 12 after initiation of steroids showing edema resolution and longitudinal lesion with pencil-thin feature at C1-C3. MRI sequences T2-weighted (a, f) and contrast-enhanced T1-weighted (b to e).
Literature review
NMOSD-like HAM patients were predominantly female (Tables 2 and S2). HAM was already present from 5 to 13 years in 17.2%. Acute onset within one month was reported in 25%, and 80.6% developed in less than 6 months. Optic or SC relapses were observed in 15.4% of cases. Clinical characteristics atypical in HAM patients were sensory level (50%), sensory or motor involvement of the upper limbs (35.9%), and optic neuritis (10.2%). Among the 4 patients with optic neuritis, one had also SC relapses, whereas among the four patients displaying SC relapses, one also had relapsing optic neuritis.
Characteristics of NMOSD-like HAM patients (literature review).

Results are given in % (n), unless otherwise stated.
aAQP4-Ab positive patients (n = 3) were excluded. MOG-Ab were not tested.
bLesions could involve cervical and dorsal levels.
cAt least from up to C3 down to T9.
Spinal MRI demonstrated extensive lesions spanning three segments or more. Lesions were purely cervical in 25.6%, extended further than the cervical region in 66.6%, and involved the whole cord in 15.4%. On T2-weighted sequences, lesions were typically linear, pencil-thin, and featuring a sagittal line (41%). Lateral funiculi were involved in 56.3% (double rope sign). Edema was observed in 51.3% and was often associated with non-specific extensive spinal lesions. Contrast enhancement was often minimal, but a thick nodular enhancement was sometimes observed along fascicular lesions. Persistent enhancement was observed during follow-up in one case.
Discussion
HAM is a slow process that progresses over years or decades, but a (sub)acute onset within the first two years is possible. In large series of HAM, acute progression was observed in 8% to 21%.5–7 The most severe patients, i.e. those with paraplegia, also had the most rapid evolution, 8 suggesting that acute inflammation is linked to severity of HAM.
The pathology of HAM lesions typically predominates on the thoracic cord, and atrophy of the lateral columns is the main autoptic finding. 9 Spinal MRI is usually normal or atrophic, and lesions are observed in fewer than 8% of patients. 8 However, MRI results acquired on historical patients were incomplete and may have long biased the design of radiological criteria of HAM in the past. Moreover, a lack of awareness of how to diagnose NMOSD, which was clearly delineated from MS only during the two last decades, and the unavailability of antibodies against AQP4 and MOG may explain the scarcity of references to NMOSD in HTLV1 infection until recently. Possible radiological myelitis in rapid progressors was not evoked before the early 2000s10,11 and is still an emerging question in the literature.
The ophthalmic signs associated with HTLV1 are mainly keratitis, uveitis, and retinal vasculitis. Optic neuritis is definitely not a typical HTLV1-induced sign. In large series of HTLV1-infected patients, none suffered from ON.12,13 However, subclinical involvement of the optic pathway may occur more often than expected in HAM patients. 14 Rare ON occurring in typical HAM or HTLV1-infected AQP4-Ab+ NMOSD patients has been reported, mostly in the Japanese literature, and incidental optic atrophy or blurred vision in HAM patients (Table S1). Unfortunately, only a few of them were examined for AQP4-Ab, and none for MOG-Ab (except our cases). The association of ON with HAM is highly suspicious of NMOSD, especially in the ancient series recruited before the delineation of NMOSD. HAM-associated uveo-papillitis is often associated with good recovery. On the other hand, our patients displayed the involvement of chiasma and residual blindness, which testifies to the severity of the ON and is suggestive of NMOSD. Unfortunately, no clinical or MRI exam was performed in patients 1 and 4 during an acute optic attack, and we only observed non-specific late optic atrophy. Although we cannot completely rule out a differential diagnosis, ON was the most plausible diagnosis and our patients were double-seronegative for AQP4 and MOG-Abs. To our knowledge, only these three patients had ON among 153 HAM or HTLV1-infected patients diagnosed in our center during the last 35 years.12,15 Based on clinical grounds, our patients were long labeled as seronegative NMOSD induced by HTLV1. However, since we finally considered that HAM might be masquerading as NMOSD, a circularity bias may have obscured this emerging diagnosis. Furthermore, it might explain the above-mentioned conflicting results in Japanese series devoid of ON, whereas multiple cases of ON or NMOSD were reported in HTLV1-infected patients.
Antibodies against AQP4 and MOG recently became available and have led to major conceptual changes regarding NMOSD over the last 15 years, including the diagnosis of atypical or incomplete NMOSD clinical pictures, and the prediction of relapses. Similar results were obtained with MOG-Ab, albeit with minor differences. Historical cases diagnosed before the availability of these antibodies remain difficult to classify, and the coexistence of a frequent infection like HTLV1 with an autoimmune disorder (NMOSD AQP4-Ab+) cannot be definitively ruled out in endemic areas. Series of symptomatic or asymptomatic HTLV1-infected patients screened for AQP4-Ab were all negative; likewise, NMOSD patients screened against HTLV1 remained negative, even in areas where both of the disorders are endemic.16–18 Therefore, the risk of misdiagnosis between NMOSD and HAM based on biological grounds is low even in high-risk populations, given that these small series included HTLV1 patients that were not selected on the basis of an acute course or MRI lesions. We therefore assume that AQP4-Ab positivity is a strong clue to real autoimmune NMOSD during fortuitous HTLV1 infection, whereas double-seronegative NMOSD-like patients probably develop clinical and imaging symptoms in relation with HTLV1 infection.
The seroprevalence of HTLV1 infection in the general population is low in endemic areas (<1% in FWI 15 ). The lifetime risk of developing HAM among HTLV1-infected patients is estimated in the range of 0.25% to 3%. 19 Therefore, assuming the independent risk of developing HAM and MS/NMOSD, only a few HTLV1-infected MS/NMOSD patients will also develop HAM, although the diagnosis can be challenging in progressive MS. Finally, we cannot exclude that the criterion of a normal cord in HAM may have induced a circularity bias, leading to the misclassification of HAM with SC lesions into MS/NMOSD categories.
The AQP4-Ab positive patients collected in this review were all characterized by acute relapsing attacks, which was a very different pattern from NMOSD-like HAM subacute onset. Therefore, although AQP4/MOG serologies were lacking in these ‘acute' HAM patients, the NMOSD-like HAM diagnosis was convincingly supported by the following: a) a consistent clinical pattern of subacute progression within a year; b) rare relapses; c) scarcity of ON; and d) suggestive MRI features. A previously unreported finding was the negative MOG and AQP4-Ab in all our patients, especially the three patients with ON history, thus supporting the diagnosis of NMOSD-like HAM in the light of the most recent criteria.
Five seronegative NMOSD-like HAM patients were collected over decades among a FWI cohort of 119 NMOSD and 153 HAM patients. HTLV1 is systematically screened in the FWI and none of the NMOSD patients was positive. We estimate that up to 4.0% (1.3 to 9.2) of NMOSD and 3.2% (1.0 to 7.2) of HAM could have been misclassified. Consequently, in populations where HTLV-1 prevalence is high, we recommend that first NMOSD attacks, especially in double seronegative patients, should be screened for HTLV-1 and specific MRI features.
This review also allowed us to characterize the MRI features of LEHAM for the first time (Table 3). Spinal cord lesions were typically pencil-thin sagittal line spanning multiple segments. Cranio-caudal edges of the lesions were poorly delimited. Rare patients demonstrated cervical lesions appearing as fragmented MS-like focal lesions or an ill-defined short sagittal line. Although our study criteria excluded myelitis shorter than three segments, rare cases supported the hypothesis of a continuum in lesion size. On axial slices, lateral funiculi (i.e. pyramidal tracts) were symmetrically involved and slightly inflated by edema, explaining the double rope sign, which is best seen on coronal slices, and the typical sagittal line lesion. Unilateral lesions were rare. Central grey matter was sometimes involved but the H-pattern was not observed. Transverse involvement of the cord was always associated with edema. Transient ill-defined swollen cervical lesions extending upward to the brainstem cortico-spinal tracts and reversed by steroids were described in two patients with HAM.20,21
Distinctive features of HAM variants and NMOSD.
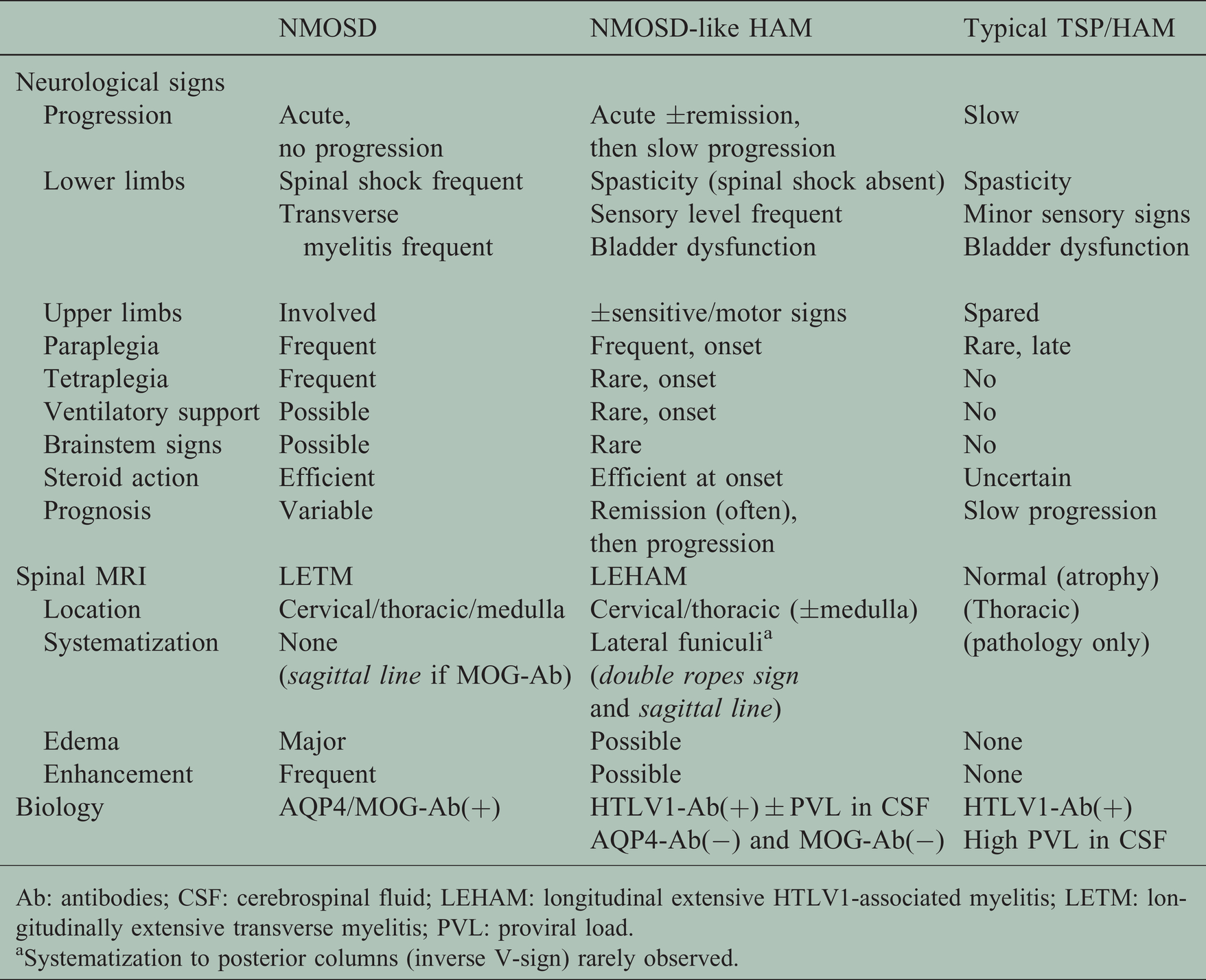
Ab: antibodies; CSF: cerebrospinal fluid; LEHAM: longitudinal extensive HTLV1-associated myelitis; LETM: longitudinally extensive transverse myelitis; PVL: proviral load.
aSystematization to posterior columns (inverse V-sign) rarely observed.
Cord swelling was long considered to be an exclusion criterion for the diagnosis of HAM, so the transient edema occurring in this series of acute HAM raises new questions. Cord edema was characteristic of acute HAM but returned to a ‘normal' size at follow-up MRI. Edema, which is a common indirect sign of inflammation, points towards an inflammation-driven acute onset of HAM, returning to a normal or atrophic size within a few months and progressing to atrophy over years. Visual assessment of SC atrophy is difficult and often leads to underestimation and lack of agreement between authors, which may explain the ‘normal' cord appearance described in disabled patients. Software calculation of the cross-sectional area of SC on images acquired with 3 T MRI demonstrated that atrophy (defined as >2SD from healthy controls) was measurable in 95.6% of HAM patients, 22 which is far higher than usually admitted in HAM. Concerning the atrophic cords, only 15.5% were limited to the thoracic cord whereas most of them also involved the cervical cord. Atrophy is known to correlate with the proportion of CD8 T cells and proviral load in CSF. 22 Interestingly, cross-sectional areas were normal at month 7 after clinical onset in two HAM patients with rapid progression. 22 MRI follow-up disclosed thoracic atrophy later extending to the rostral cord, which was interpreted as retrograde Wallerian degeneration of the cervical cord. Although it has been reported only in extreme cases, this transient inflammation suggests that diffuse subtle cord edema at onset might go undetected at the onset of HAM owing to the pseudo-normal cord size. Contrast uptake was mostly minor or absent. Some cases were enhanced along the funiculi with a linear or patchy, usually symmetrical aspect.
The following patterns were not observed in LEHAM: MS-like sub-ependymal focal well-limited lesion; isolated conus lesion, except in association with holocord involvement; heterogenous patchy irregular cord edema; central necrosis or pseudo-syringomyelia, as observed in NMOSD. The main differential diagnosis of HAM sagittal lesions was MOG-associated disorder (MOGAD),23,24 but axial slices of MOGAD typically demonstrate bright spotty lesions or H-lesions involving grey matter, which were not observed here. Severe B12 deficiency predominates in the posterior column (inverted V sign), and rare mitochondrial disorders combine the double rope sign with flourish lesions. 25 Very rare paraneoplastic disorders, mostly CRMP5-Abs, may target the lateral funiculi, 26 but the association with cancer, acute onset and poor outcome is different from our cases. To our knowledge, the double rope sign has never been observed before.
The outcome of the cervical or thoracic lesions was their complete regression on MRI within a year, although normalization of MRI does not necessarily mean clinical improvement. Small residual T2w lesions were evidenced in rapidly progressing patients, suggesting that early acute cervical/thoracic myelitis may have been missed by late-acquired MRI. Since our HAM patients were not followed up as closely as MS or NMOSD patients, transient exacerbation or inflammation might be easily missed by late-acquired images. The natural history of SC lesions in HAM is still unclear, and the regular follow-up of patients during the early phase of the disease may lead to new findings that could raise therapeutic issues.
Finally, the natural history of MRI myelopathy in HAM suggests the succession of two phases 27 (Figure 3):
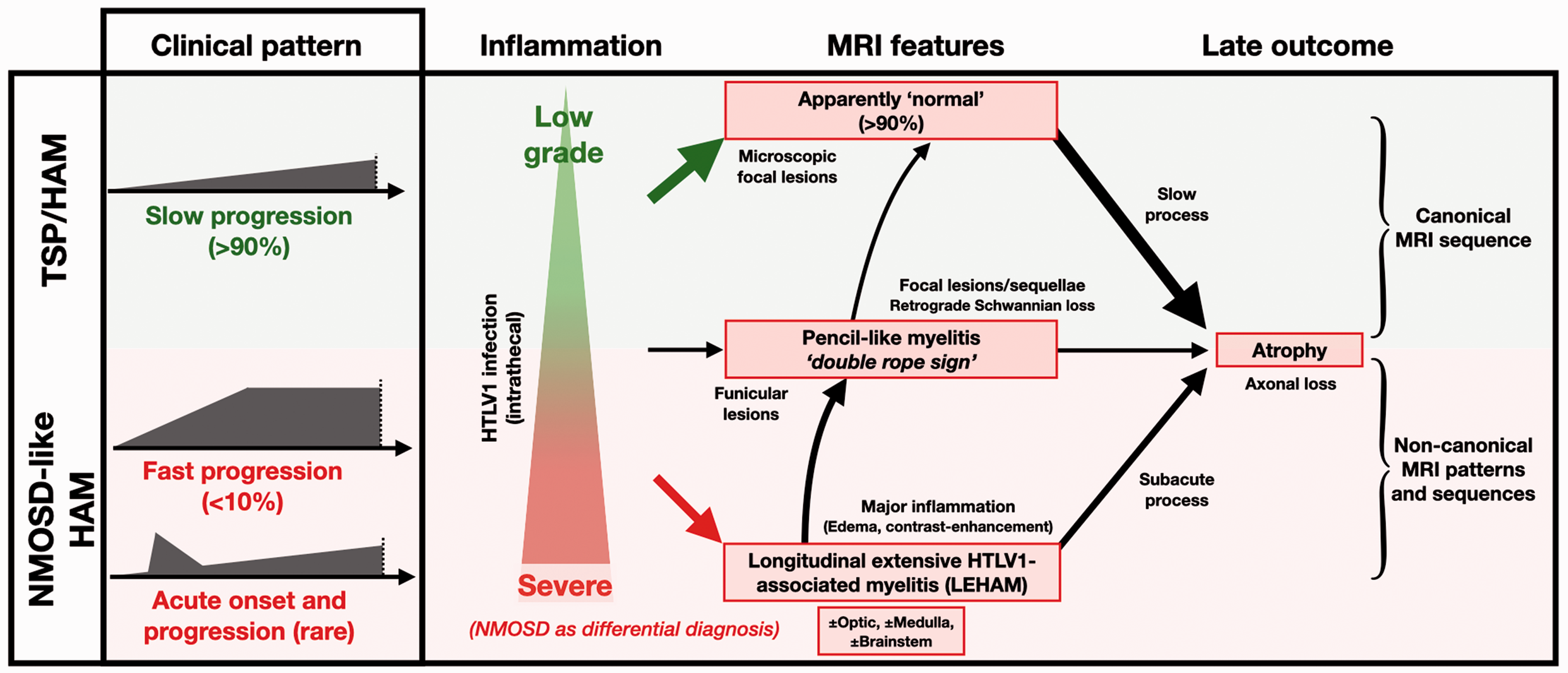
Clinical and MRI sequences during HTLV1-associated myelitis (HAM). Classical tropical spastic paraparesis (TSP)/HAM is a slowly progressive myelopathy evolving over years or decades. Spinal cord MRI is normal at onset and slowly progresses to atrophy (canonical MRI sequence). A minority of TSP/HAM patients may undergo fast progression (over weeks or months), or even feature NMOSD attacks. Clinical signs may extend to cervical and thoracic cord, optic nerve, medulla and brainstem, and may lead to severe impairment. Spinal MRI demonstrates longitudinal extensive HAM (LEHAM) with edema and contrast enhancement. Remission of edema and enhancement is obtained within weeks or months and gives way to apparently ‘normal' cord or atrophy. Prevalence of NMOSD-like HAM onset remains low but could be underestimated. Differential diagnosis with double AQP4/MOG-Ab NMOSD is raised in these non-canonical MRI patterns of HAM. Besides the two extreme presentations, high-quality MRI may show pencil-thin myelitis limited to pyramidal tracts (double rope sign), featuring a continuum of MRI lesions extending from normal profile to longitudinal extensive transverse myelitis.
an acute inflammatory phase characterized by an abrupt clinical onset over weeks or months, severe impairment, extensive myelitis, and cord edema (LEHAM). This optional phase is partially sensitive to steroids, cord MRI improves within months, but clinical impairment may remain.
classical chronic myelopathy, associated with slowly progressive impairment and cord atrophy without overt inflammation on MRI.
Although the severe acute inflammatory phase was rarely evidenced by MRI, the description of transitional patients showing LEHAM without acute clinical onset is probably the macroscopic translation of microscopic inflammation of the lateral funiculi triggered by HTLV1 infection, given the highly variable severity of the underlying inflammation. Future longitudinal studies of spinal MRI acquired in early diagnosed HAM might clarify this issue.
Although chronic myelopathy is the rule in HAM, acute onset with myelitis is an extreme on the HAM spectrum and the diagnosis of acute episodes could be challenging. This review suggests that extensive spinal lesions are associated with atypical clinical characteristics, which are classically considered as evidence against the diagnosis of HAM. Sensory level and constricting pain of the abdomen are commonly reported in these patients and are usually reversed by steroid pulses. According to the cervical level of the lesions, the upper limbs are often affected by sensory or pyramidal signs, whereas HAM is commonly restricted to the lower limbs and abdomen.
Spasticity is always a prominent early feature of HAM, and the term tropical spastic paraparesis (TSP) is well coined. Flaccid paraplegia was not observed in patients with HAM myelitis, pointing to a major clinical difference with NMOSD. The latter was sometimes associated with extensive spinal necrosis with neurons and peripheral motor nerve lesions, and acute spinal shock was associated with the prognosis. Severe spasticity as observed in HAM or MS is highly uncommon in the bedside experience of NMOSD, although the prevalence and severity of spasticity in NMOSD patients deserve future studies. In extensive myelitis with minor symptoms, the association of spasticity, urinary disturbance, and sparing of the sensory system should point to HAM rather than NMOSD. Lastly, the systematic presence of myelitis lesions on the funiculi strongly supports the diagnosis of LEHAM rather than NMOSD.
In a previous study, we found that proviral load was always positive in the CSF of HAM patients and that high proviral load correlated with rapid progression. 28 We cannot exclude the possibility that the CSF proviral load required to trigger acute inflammation may not have reached the detection limit at the time of onset, or the possibility that the pathophysiologic mechanisms may have shifted before the CSF positivity was detected. Lastly, the influence of proviral load on acute progression is an indirect sign, since proviral load was obtained late after acute onset, 6 which may not mirror the proviral load in the acute phase and simply provides a retrospective explanation. It is possible that the lower proviral load obtained in our two patients indicates a different pathogenesis in acute onset patients, and that the predictive capacity of results obtained in progressive patients are not applicable in NMOSD-like HAM patients. The follow-up of proviral load would have been informative but was not obtained in our patients.
Our study has several limitations. First, proviral loads were not always available. Second, clinical and biological data and the MRI quality of cases from the literature review were incomplete and heterogeneous, preventing any statistical analysis, and we could only estimate the prevalence of LEHAM signs. On the other hand, this is the first time a large series of acute-onset HAM sharing features with NMOSD has been analyzed, with emphasis on a typical pattern of lesions. In addition, the double seronegativity of our series, which was obtained with recent sensitive techniques, provides further evidence for the direct responsibility of HTLV1 instead of NMOSD. In our opinion, the radiological features of HAM may henceforth be envisaged as covering a wider spectrum of features ranging from an apparently normal spinal cord to NMOSD-like myelitis, according to the clinical profile. Furthermore, a prospective study of HAM patients included at onset should now be conducted. However, the epidemiological trend of HAM in the FWI has tended to a null incidence over the last decade, 15 which is a definitive limitation for further local studies.
While HTLV1 infection may mimic NMOSD attacks including their response to steroids, LEHAM is a new differential diagnosis of LETM and HTLV1 serology should be performed in double (AQP4/MOG)-seronegative patients from high-risk areas, especially in the presence of red flags.
Supplemental Material
sj-pdf-1-mso-10.1177_20552173211037361 - Supplemental material for NMOSD-like and longitudinal extensive HTLV1-associated myelitis are extremes that flank an overlooked continuum
Supplemental material, sj-pdf-1-mso-10.1177_20552173211037361 for NMOSD-like and longitudinal extensive HTLV1-associated myelitis are extremes that flank an overlooked continuum by Mickael Bonnan Stéphane Olindo Quentin Lobjois Dalia Dimitri Boulos Philippe Cabre in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-pdf-2-mso-10.1177_20552173211037361 - Supplemental material for NMOSD-like and longitudinal extensive HTLV1-associated myelitis are extremes that flank an overlooked continuum
Supplemental material, sj-pdf-2-mso-10.1177_20552173211037361 for NMOSD-like and longitudinal extensive HTLV1-associated myelitis are extremes that flank an overlooked continuum by Mickael Bonnan Stéphane Olindo Quentin Lobjois Dalia Dimitri Boulos Philippe Cabre in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-pdf-3-mso-10.1177_20552173211037361 - Supplemental material for NMOSD-like and longitudinal extensive HTLV1-associated myelitis are extremes that flank an overlooked continuum
Supplemental material, sj-pdf-3-mso-10.1177_20552173211037361 for NMOSD-like and longitudinal extensive HTLV1-associated myelitis are extremes that flank an overlooked continuum by Mickael Bonnan Stéphane Olindo Quentin Lobjois Dalia Dimitri Boulos Philippe Cabre in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Supplemental Material
sj-pdf-4-mso-10.1177_20552173211037361 - Supplemental material for NMOSD-like and longitudinal extensive HTLV1-associated myelitis are extremes that flank an overlooked continuum
Supplemental material, sj-pdf-4-mso-10.1177_20552173211037361 for NMOSD-like and longitudinal extensive HTLV1-associated myelitis are extremes that flank an overlooked continuum by Mickael Bonnan Stéphane Olindo Quentin Lobjois Dalia Dimitri Boulos Philippe Cabre in Multiple Sclerosis Journal – Experimental, Translational and Clinical
Footnotes
Acknowledgements
We are indebted to Ray Cooke for copyediting the manuscript.
Contributorship
MB, PC were responsible of study concept and design, data acquisition, and radiographic data review. MB interpreted the data and drafted of manuscript. All authors provided a critical review and approved the final manuscript. PC is the guarantor.
Ethic approval
This study fulfils French law controlling non-interventional retrospective studies using de-identified data, and was approved by the local ethic committee.
Conflict of Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data Accessibility Statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Supplemental material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.