
Abstract
Background
Contactin-1 and contactin-2 are important for the maintenance of axonal integrity.
Objective
To investigate the cerebrospinal fluid levels of contactin-1 and contactin-2 in multiple sclerosis patients and controls, and their potential use as prognostic markers for neurodegeneration.
Methods
Cerebrospinal fluid contactin-1 and contactin-2 were measured in relapsing–remitting multiple sclerosis (n = 41), secondary progressive multiple sclerosis (n = 26) and primary progressive multiple sclerosis patients (n = 13) and controls (n = 18), and in a second cohort with clinically isolated syndrome patients (n = 88, median clinical follow-up period of 2.3 years) and controls (n = 20). Correlations/linear regressions were analysed with other baseline cerebrospinal fluid axonal damage markers and cross-sectional/longitudinal magnetic resonance imaging features.
Results
Contactin-1 and contactin-2 levels were up to 1.4-fold reduced in relapsing–remitting multiple sclerosis (contactin-1: p = 0.01, contactin-2: p = 0.02) and secondary progressive multiple sclerosis (contactin-1: p = 0.05, contactin-2: p = 0.02) compared to controls. In clinically isolated syndrome patients, contactin-1 tended to increase when compared to controls (p = 0.07). Both contactin-1 and contactin-2 correlated with neurofilament light, neurofilament heavy and magnetic resonance imaging metrics differently depending on the disease stage. In clinically isolated syndrome patients, baseline contactin-2 level (β = –0.42, p = 0.04) predicted the longitudinal decline in cortex volume.
Conclusion
Cerebrospinal fluid contactin-1 and contactin-2 reveal axonal dysfunction in various stages of multiple sclerosis and their inclusion to the biomarker panel may provide better insight into the extent of axonal damage/dysfunction.
Introduction
There is increasing evidence that neurodegeneration through axonal damage or dysfunction occurs in the early course of multiple sclerosis (MS) independent of demyelination, and is responsible for irreversible neurological disability.1,2 In the past few decades, there has been growing interest in developing cerebrospinal fluid (CSF) biomarkers that reflect axonal loss or damage in the early stages of MS.3–6 Biomarkers such as neurofilaments, tau, 14–3-3 and N-acetyl aspartic acid might be helpful to detect and monitor the extent of axonal destruction.6–8 However, in view of the axonal damage present in the early disease stage that is likely to be preceded by axonal dysfunction, markers that reflect axonal dysfunction are needed. Ultimately, a specific signature from a combination of these CSF axonal damage and dysfunction proteins will pave the way for improved prognostic accuracy in MS and better monitoring of response to therapies.
Contactin-1 and contactin-2 are brain-specific soluble cell adhesion proteins of the contactin family, expressed on the axonal membranes of neurons,9,10 and are suggested to reflect axonal dysfunction. These two proteins have a similar structure and both interact with a well-known protein involved in axonal damage, i.e. amyloid precursor protein,11,12 but they differ in their axonal localisation. Contactin-1 is expressed in paranodal axonal domains, whereas contactin-2 is localised in the juxtaparanode.13,14 These proteins may therefore play different roles in the pathology of MS. Contactin-1 has been reported to be involved in myelin formation in the central nervous system (CNS) by way of axo-glia interaction, the loss of which is one of the main causes of neuronal dysfunction in MS. 15 Indeed, in chronic MS lesions, contactin-1 was highly expressed in demyelinated axons, probably to induce remyelination. 16 Contactin-2 was found to be involved in axonal growth and guidance. 17 It was recently identified as one of the elevated CSF proteins in a proteomics study in paediatric MS patients compared to those with monophasic CNS demyelinating syndrome 18 and in clinically isolated syndrome (CIS) patients versus controls. 19
Based on their role in axonal domain organisation,13,14 we hypothesised that CSF levels of contactin-1 and contactin-2 are altered differently in various MS subtypes and may serve as surrogate markers for early axonal domain dysfunction. Contactin-1 and contactin-2 were measured at baseline in the CSF of patients with different subtypes of MS, as well as in patients with early MS (CIS) that progressed to the diagnosis of clinically definite multiple sclerosis (CDMS) at follow-up. We investigated the relationship of contactin-1 and contactin-2 with axonal damage markers such as neurofilament light (NFL), neurofilament heavy (NFH) and brain atrophy markers at baseline in all MS subtypes as well as with longitudinal change in cortex volume in CIS patients.
Materials and methods
Subjects
We included patients from two cohorts: the MS outpatient clinic of the VU University Medical Centre, Amsterdam, The Netherlands (cohort 1) and the Department of Neurology/Medical University of Graz, Graz, Austria (cohort 2).
Cohort 1 consisted of MS patients (n = 80) and controls (n = 18, in which 14 subjects had non-inflammatory neurological disease such as paraparesis (non-progressive), vision problems (strabismus), headache, non-inflammatory polyneuropathy and young stroke protocol; and four subjects had inflammatory neurological disease such as PML, herpes infection and cerebral vascular pathology) who volunteered to participate in the study after an advertisement. MS patients in cohort 1 were diagnosed according to the state-of-the-art diagnostic criteria when CSF was collected (between 2000 and 2005),20, 21 and further classified according to Lublin and Reingold diagnostic criteria 22 as having relapsing–remitting multiple sclerosis (RRMS) (n = 41), secondary progressive multiple sclerosis (SPMS) (n = 26) or primary progressive multiple sclerosis (PPMS) (n = 13). CSF was collected during remission, except for two patients who experienced a relapse in the month preceding CSF collection. The minimum time period was 6 weeks after a relapse to consider that a patient was in remission at the time of CSF collection. The average time from disease onset to CSF collection was 9.5 ± 9 years. No patients were treated with methylprednisolone.
Cohort 2 consisted of patients with CIS (n = 88) and controls (n = 20). CSF samples were collected between 2003 and 2010. 8 Patients were selected when they fulfilled the criteria (Supplementary Methods section 1). CIS patients (n = 79), were classified as either CDMS (n = 35) and patients who did not convert to MS (n = 44) after a clinical follow-up period of 2.3 (1.4–3.4) years (median (interquartile range; IQR)). The follow-up details of nine patients were not available for cohort 2. Therefore, they were excluded from the follow-up analyses. Control group subjects in both the cohorts were identified using the consensus criteria for control groups from the BioMS consortium. 23 The demographic and clinical details of all patients are outlined in Table 1. This study was performed in agreement with the ethical principles of the Declaration of Helsinki. The ethical review boards of the VU University Medical Centre and Medical University of Graz approved the cohorts and all subjects provided written informed consent.
Demographic and clinical details of patients.
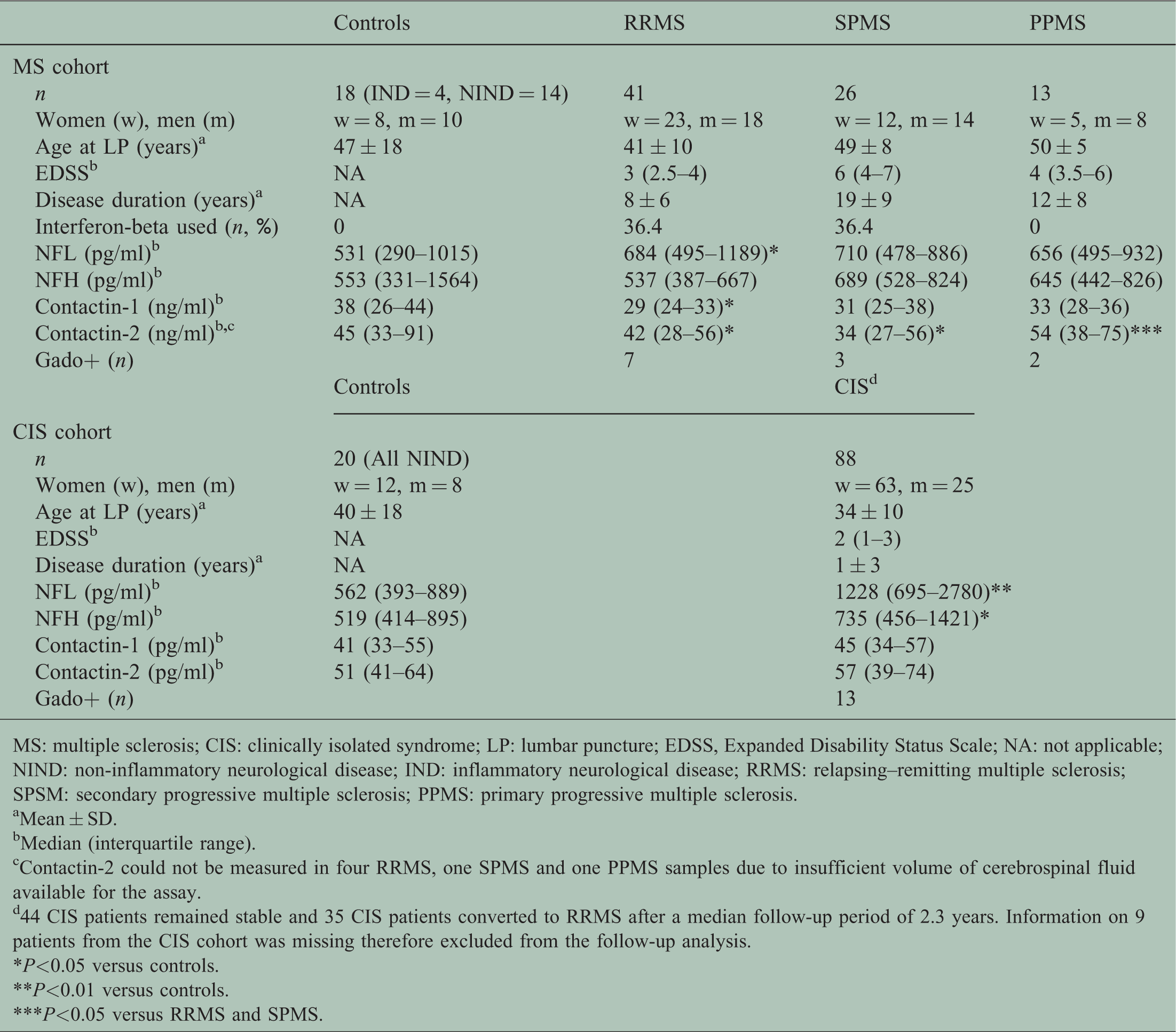
MS: multiple sclerosis; CIS: clinically isolated syndrome; LP: lumbar puncture; EDSS, Expanded Disability Status Scale; NA: not applicable; NIND: non-inflammatory neurological disease; IND: inflammatory neurological disease; RRMS: relapsing–remitting multiple sclerosis; SPSM: secondary progressive multiple sclerosis; PPMS: primary progressive multiple sclerosis.
Mean ± SD.
Median (interquartile range).
Contactin-2 could not be measured in four RRMS, one SPMS and one PPMS samples due to insufficient volume of cerebrospinal fluid available for the assay.
44 CIS patients remained stable and 35 CIS patients converted to RRMS after a median follow-up period of 2.3 years. Information on 9 patients from the CIS cohort was missing therefore excluded from the follow-up analysis.
*P<0.05 versus controls.
**P<0.01 versus controls.
***P<0.05 versus RRMS and SPMS.
CSF samples and enzyme-linked immunosorbent assay analysis
CSF was collected by standard lumbar puncture and stored according to guidelines until analysis. 4 Commercially available enzyme-linked immunosorbent assays (ELISAs) were used for measuring contactin-1 (RayBiotech, USA) and contactin-2 (Contactin-2 duoset ELISA kit; R&D, Minneapolis, USA) in the CSF (details of the ELISAs used in this study are given in Supplementary Methods section 2). All analyses were performed blinded to the patient details.
Magnetic resonance imaging
In Amsterdam (cohort 1), magnetic resonance imaging (MRI) examination was performed within 3 weeks of CSF collection. The T2 lesion load was quantified using home-developed semi-automated seed-growing software based on a local thresholding technique (Supplementary Methods section 3). To assess brain atrophy, normalised brain volumes were measured using structural image evaluation, using normalisation of atrophy cross-sectionally (SIENAX).24,25
In Graz (cohort 2), all patients underwent MRI of the brain at baseline on a 3T Tim Trio system (Siemens Medical Systems, Erlangen, Germany) using a 12-element phased-array head coil, and further processing was carried out according to our previous study. 8 The follow-up MRI protocol was identical to the baseline protocol. For assessing the T2 lesion load, MS lesions were manually segmented using a region growing algorithm that was based on local thresholding. The normalised cortical volume at baseline and follow-up was assessed with focussed abdominal sonography for trauma (FAST) from the FMRIB Software Library (FMRIB, Oxford, UK). Changes in brain volume over time were measured with SIENA25,26 from the same library.
Statistics
Statistical analyses were performed using SPSS version 22 (IBM SPSS Statistics for Windows, version 21.0; IBM Corp., Armonk, NY, USA). Graphs were plotted using GraphPad Prism version 6.07.
Contactin-1 and contactin-2 were found to be normally distributed. NFL and NFH were log-transformed for normal distribution. Differences in CSF contactin-1, contactin-2, NFL and NFH levels between two diagnostic groups (MS patients combined (RRMS/SPMS/PPMS) vs. controls) were analysed by general linear models adjusted for age and/or sex (when association with age and/or sex was found). Multiple group comparisons (among four groups RRMS, SPMS, PPMS and controls) were similarly performed using analysis of covariance adjusted for age and/or sex (when applicable). Corrections for multiple comparisons were not applied. Correlation analyses were performed using Spearman’s test or Pearson’s partial correlation test (when age correction was applied). A multiple stepwise linear regression was performed to predict the change in cortex volume during follow-up based on CSF axonal damage markers (contactin-1, contactin-2, NFL and NFH). The statistical tests were two-tailed and values with p<0.05 were considered significant.
Results
Correlations with demographic data in CDMS and group differences
CSF contactin-1 (p = 0.01, adjusted for age, Figure 1(a)) and contactin-2 (p = 0.02, Figure 1(b)) were decreased in MS patients compared to controls. Contactin-1 [F(3,93) = 2.29; p = 0.08, adjusted for age] tended to differ and contactin-2 [F(3,90) = 3.43; p = 0.02] levels differed among control, RRMS, SPMS and PPMS groups. Contactin-2 was not correlated with age (r = 0.03, p = 0.8), thus no age correction was applied. Post-hoc analysis revealed that contactin-1 levels were decreased in RRMS (p = 0.02, Figure 1(a)) and SPMS (p = 0.03, Figure 1(a)) versus controls. Contactin-2 levels were likewise reduced in both RRMS (p = 0.02, Figure 1(b)) and SPMS (p = 0.01, Figure 1(b)) compared to controls. Moreover, contactin-2 levels were higher in PPMS than RRMS (p = 0.06, Figure 1(b)) and SPMS (p = 0.04, Figure 1(b)) patients and overall lower in relapse-onset MS (RRMS and SPMS combined) than PPMS (p = 0.03). Both contactin-1 and contactin-2 did not correlate with the Expanded Disability Status Scale within any diagnostic group (data not shown).

Levels of cerebrospinal fluid contactin-1 (a) and contactin-2 (b) in controls and multiple sclerosis patients. Each dot in the scatter box-plot represents a sample. The long horizontal line represents median and the short horizontal lines represent interquartile range, respectively.
Correlations with levels of CSF biomarkers for axonal damage in MS patients
In RRMS patients, contactin-1 correlated with NFH (r = 0.40, p = 0.01, Figure 2(b)). In SPMS patients, contactin-1 correlated with both NFL (r = 0.48, p = 0.01, Figure 2(e)) and NFH (r = 0.46, p = 0.02, Figure 2(f)). In PPMS patients, contactin-1 did not correlate with NFL (r = 0.51, p = 0.07) but correlated with NFH (r = 0.62, p = 0.03) (see Supplementary Figure 1). The correlations remained significant after correcting for age except in the PPMS group. CSF contactin-2 levels did not correlate with NFL or NFH levels within the RRMS (Figure 2(c) and 2(d)), SPMS (Figure 2(g) and 2(h)) and PPMS groups (Supplementary Figure 1). In control subjects, no correlations between contactin-1 or contactin-2 and NFL or NFH were observed.
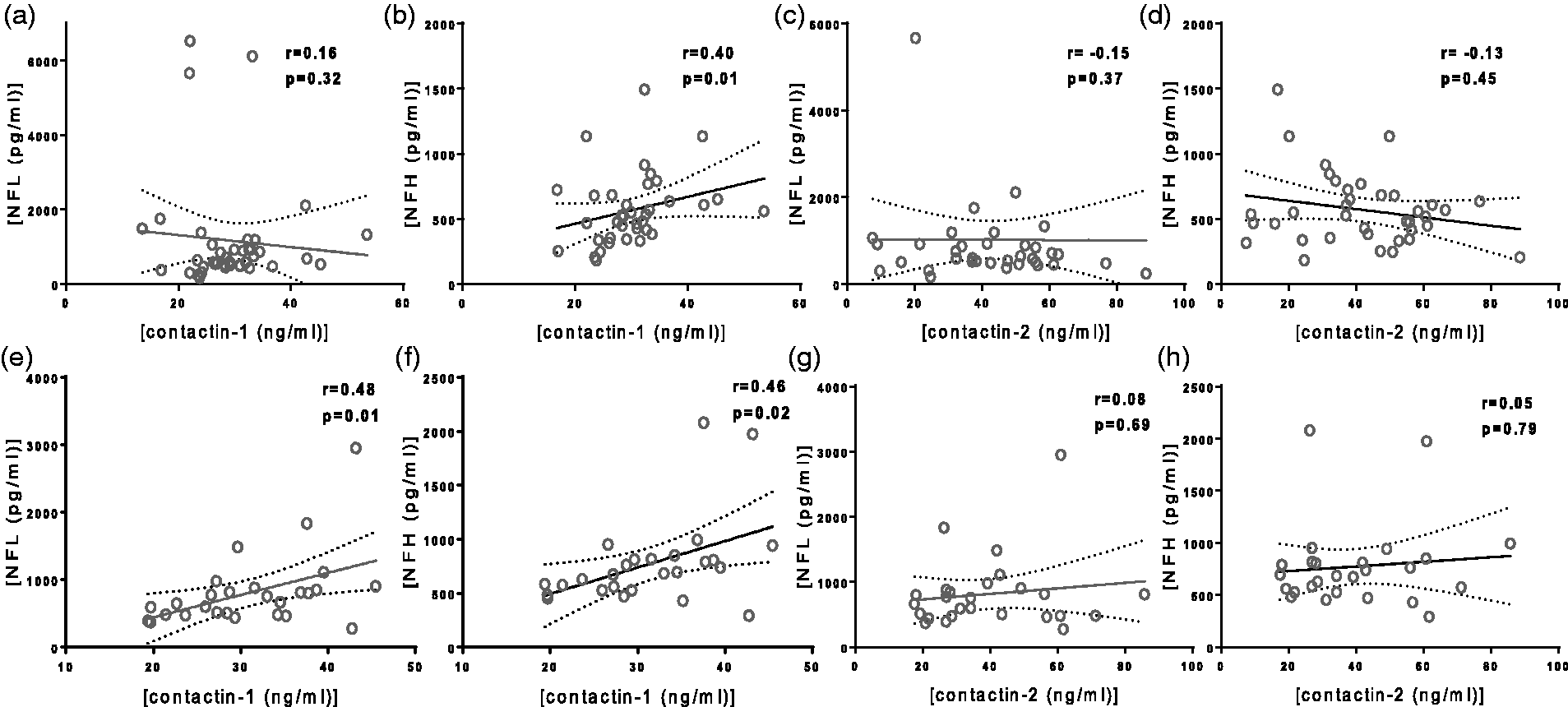
Correlation of cerebrospinal fluid (CSF) contactin-1 (a,b,e,f) and contactin-2 (c,d,g,h) with CSF neurofilament light and neurofilament heavy in relapsing–remitting multiple sclerosis (top panel) and secondary progressive (bottom panel) multiple sclerosis patients. Each dot in the scatter plot represents a sample. r is Spearman’s correlation coefficient.
Correlations with brain tissue damage in MS patients
In SPMS patients, contactin-1 positively correlated with normalised brain volume (r = 0.38, p = 0.05, Figure 3(a)) and negatively correlated with T2 lesion load (r = –0.39, p = 0.04, Figure 3(b)). Within the RRMS and PPMS groups, no correlations between contactin-1 and contactin-2 and normalised brain volume or T2 lesion load were found. However, in combined MS group, contactin-2 positively correlated with normalised brain volume (Supplementary Figure 2).

Correlation of cerebrospinal fluid contactin-1 with imaging biomarkers – normalised brain volume (a) and T2 lesion load (b) in secondary progressive multiple sclerosis patients. Each dot in the scatter plot represents a sample. r is Spearman’s correlation coefficient.
Group differences and correlations with demographic data in CIS
In contrast to the lower levels in MS patients, CSF contactin-1 levels tended to increase in CIS patients (p = 0.07, data adjusted for age and sex, Figure 4(a)) compared to controls. CSF contactin-2 levels were comparable in CIS patients (p = 0.15, adjusted for sex, Figure 4(b)) and controls (see Supplementary Figure 4). Furthermore, baseline CSF contactin-1 and contactin-2 levels were similar in CIS patients who remained stable versus CIS patients who converted to CDMS (median follow-up (years) 2.3 (1.4–3.4) (see Supplementary Figure 5)). However, contactin-1 was higher (p = 0.03) in stable CIS patients than controls.

Levels of cerebrospinal fluid contactin-1 (a) and contactin-2 (b) in controls and patients with clinically isolated syndrome. Each dot in the scatter box-plot represents a sample. The long horizontal line represents median and the short horizontal lines represent interquartile range.
Correlations with levels of CSF NFL and NFH in CIS
Contactin-1 did not correlate with NFH (r = 0.19, p = 0.07, Figure 5(a)), while contactin-2 correlated weakly with NFH (r = 0.23, p = 0.04, Figure 5(b)). No correlations with NFL were observed in CIS patients. In controls, after age correction, only contactin-1 was found to correlate positively with NFL (r = 0.60, p = 0.02).
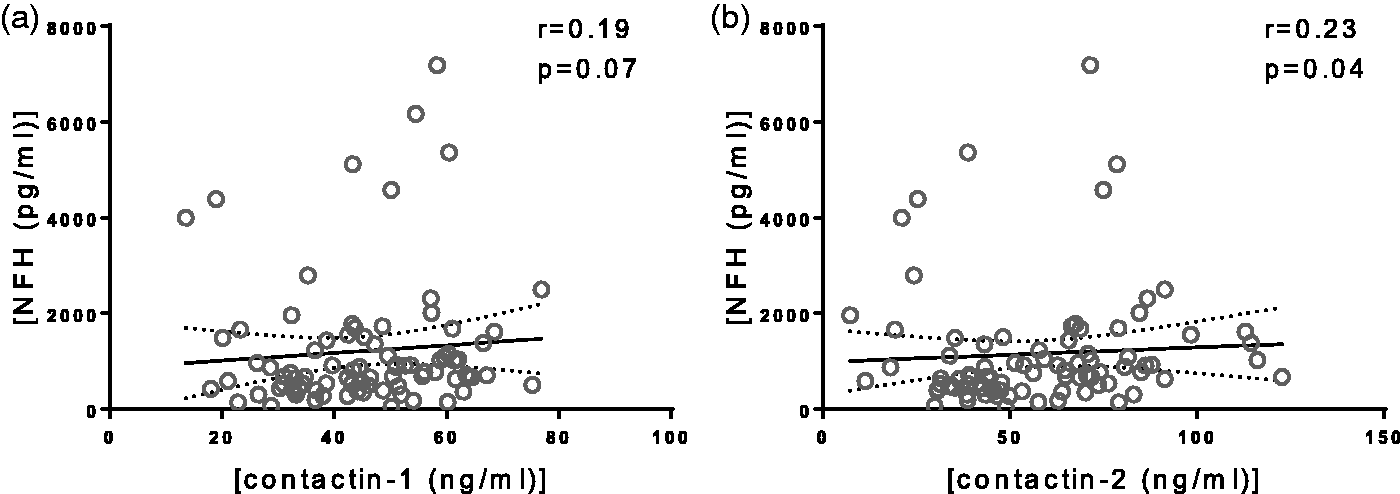
Correlation of cerebrospinal fluid (CSF) contactin-1 (a) and contactin-2 (b) with CSF neurofilament heavy in clinically isolated syndrome patients. Each dot in the scatter plot represents a sample. r is Spearman’s correlation coefficient.
Association with brain tissue damage in CIS patients
There were no correlations of CSF contactin-1 and contactin-2 with baseline normalised cortex volume and T2 lesion load (data not shown). Interestingly, only contactin-2 [F(1,22) = 4.63, p = 0.043, standardised β = –0.417, R2 = 0.174, Table 2] could predict the longitudinal change in cortex volume. The correlation between contactin-2 and the change in cortex volume is shown in Supplementary Figure 6. None of the other variables (contactin-1, NFL, NFH) were found to be significant predictors of brain atrophy over time in CIS patients.
Output of linear regression.
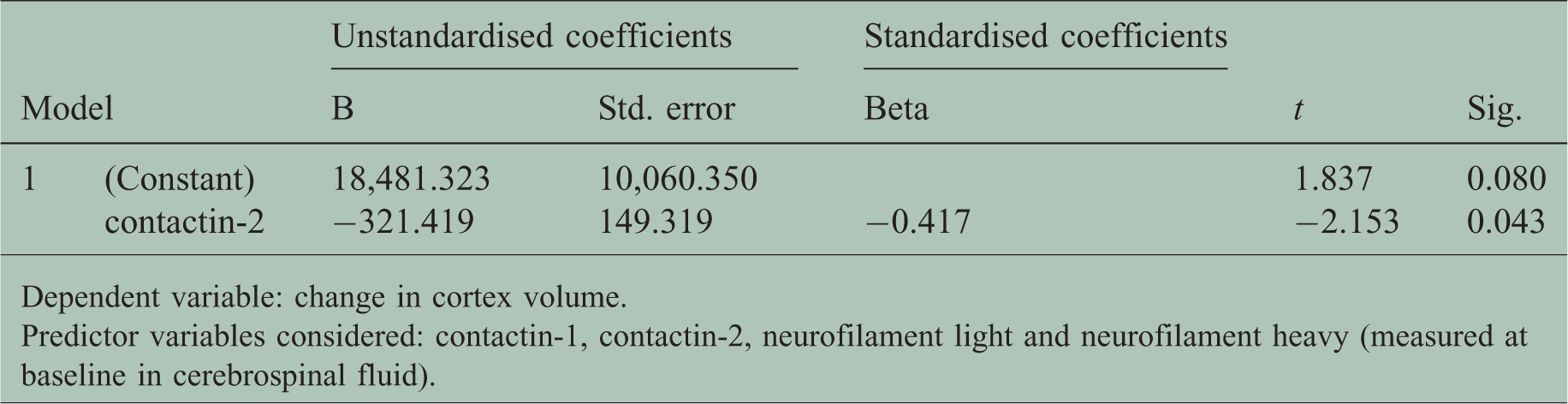
Dependent variable: change in cortex volume.
Predictor variables considered: contactin-1, contactin-2, neurofilament light and neurofilament heavy (measured at baseline in cerebrospinal fluid).
Discussion
In this study, we measured the levels of contactin-1 and contactin-2 in CSF, first in CDMS patients, and next in patients with CIS, to evaluate the potential of these proteins as markers for axonal domain dysfunction. Our results suggest that the reduction of CSF levels of both contactin-1 and contactin-2 in RRMS and SPMS reflect the underlying axonal pathology in these disease subtypes. In contrast to definite MS patients, contactin-1 tended to increase in patients with CIS compared to controls. In addition, contactin-2 was the most significant predictor of longitudinal brain atrophy in CIS patients.
Despite the small sample size of our MS cohort, we observed decrements in contactin-1 and contactin-2 levels in RRMS and SPMS compared to controls, which were not found in case of NFL and NFH (Table 1). Our finding that contactin-1 is reduced in RRMS is in accordance with proteomics studies that found contactin-1 among the proteins that were reduced in the CSF of RRMS patients compared to controls.27,28 However, our finding of decreased levels in SPMS compared to controls was in contrast to the findings of a proteomics study that found increased contactin-1 levels in SPMS compared to controls.27,28 This discrepancy could be due to different measurement platforms (proteomics used in previous studies vs. ELISA used in our study) and possibly different epitopes of the antibodies. Moreover, contactin-1 associates with sodium channels and in particular, Nav1.2, enhancing the surface expression of the latter. 29 Diffuse high axonal expression of Nav1.2 has been found in MS plaques. 30 Therefore, by modulating the surface expression of Nav1.2, contactin-1 may contribute to a putative compensatory mechanism to restore axonal function in early MS, which might lead to the decreased release of contactin-1 in the CSF and thus lower levels in MS. Alternatively, some studies31,32 have shown axonal degeneration and significant reduction of axonal density in SPMS. Similar reduced levels of axonal protein have been observed earlier in SPMS, such as N-acetyl-aspartate, which supports decreases in axonal biomarkers with disease progression. 33 Based on these previous studies, we hypothesised that significantly lower levels of contactins in SPMS compared to controls could reflect slow axonal degeneration in the progressive phase. Thus, contactin-1 could maybe even be a marker of evolution to a secondary phase. In contrast to RRMS and SPMS, even though not highly significant, slightly higher levels of both contactin-1 and contactin-2 in PPMS might suggest a different mechanism. In a progressive stage such as PPMS, the active process of axonal loss could lead to significantly higher levels of axonal proteins such as tau and MOG in CSF. 34 A similar process might lead to slightly higher levels of contactin-1 and contactin-2 in the CSF of a subset of PPMS patients.
Our finding that contactin-1 tended to increase in CIS is in agreement with a previous proteomics study in which contactin-1 levels were found to be higher in CIS than controls. 35 However, baseline contactin-1 and contactin-2 levels were similar in CIS patients who remained stable and converted to RRMS at the last visit. Slightly higher contactin-1 levels in CIS could be due to acute release during axonal myelin domain dysfunction, or a restorative mechanism.
We next studied the relationship of contactins with neurofilaments (biomarkers for axonal damage), which has not been investigated so far. We found that contactin-1 correlated positively with NFH in CIS, RRMS and SPMS patients. The pattern of alterations of contactin-1 and NFH are similar between various MS subtypes, which possibly explains the observed positive correlations of contactin-1 with NFH (see Supplementary Figure 3). Although the sample size of the PPMS group was small, we found a correlation between contactin-1 and NFH within this group Supplementary Figure 1). In contrast, contactin-1 correlated with NFL only in progressive MS patients, i.e. SPMS and PPMS (r = 0.45, p = 0.004). These results indicate that contactin-1 is related to NFH and NFL differently depending on the disease stage and therefore likely reflects a different extent of axonal damage.
Contactin-1 and contactin-2 did not correlate with brain atrophy markers in CIS and RRMS patients in cross-sectional analyses. However, contactin-1 correlated positively with normalised brain volume in SPMS patients, implying that decreased brain volume or atrophy is reflected by reduced CSF levels of contactin-1. In addition, contactin-1 correlated negatively with the T2 lesion load in SPMS patients, suggesting that reduced contactin-1 levels may be associated with increased lesion load in the progressive disease stage. In CIS patients, contactin-2 was negatively associated with the change in cortex volume during follow-up and was the best predictor of brain atrophy among contactin-1, NFL and NFH. These results indicate the possible use of contactin-2 as a biomarker for monitoring disease progression, as the baseline levels could predict the subsequent cortical volume changes. Larger studies with longer follow-ups must confirm whether the relation of contactin-2 with atrophy is indeed only present in CIS patients and disappears in definite MS. Overall, the correlations of contactins with MRI, which is considered one of the most powerful techniques for the differential diagnosis of MS, further support the idea that contactin levels might also reflect neuronal damage or brain atrophy in MS.
The major strengths of our study were that we used analytically validated commercially available ELISAs for measuring contactin levels, which is an advantage over previous studies involving contactins analysed by proteomics methods. The commercial availability facilitates replication of the findings. In addition, we used a larger sample size compared to previous studies. We included two independent cohorts from two geographical locations, which extended the assessment of suitability of contactin-1 and contactin-2 as markers for axonal domain dysfunction in different stages of MS. Nevertheless, our study has some limitations. First, the control group of cohort 1 was small and heterogeneous. However, the mix of inflammatory and non-inflammatory disease represents a real clinical setting. As not a lot of studies have investigated contactins in MS, our study aimed to explore the levels of contactins in all subtypes of MS as well as in inflammatory and non-inflammatory cases that come to the MS centre. As there is not much information yet on contactin-1 and contactin-2 levels in other neurological diseases, we cannot exclude that they are altered in those diseases. This novel MS biomarker study was exploratory in nature and future large cohort studies are warranted to evaluate the value of contactins as diagnostic/prognostic biomarkers. Second, we were limited to cross-sectional data in the definite MS cohort, which restricted our statistical analysis method to correlations. Third, due to lack of follow-up CSF, the differences in the levels of contactins in CIS patients who converted to CDMS and those patients who remained stable could not be analysed after the follow-up time point. As the CIS and RRMS patients were from different cohorts from two distinct locations, we could not compare contactin-1 and contactin-2 levels or imaging-derived metrics between these two groups directly. Although we found that both contactin-1 and contactin-2 baseline levels were similar in CIS patients converting to CDMS versus CIS patients who remained stable (cohort 2), the sample size was small for a statistically robust comparison. Moreover, IQR for follow-up time in this cohort starts at 1.4 years, which may be insufficient follow-up time for some patients to convert to MS. Future studies should include larger sample sizes for comparisons of CIS converting into CDMS versus stable CIS patients, and should explore the possibility of contactin-1 or contactin-2 to predict conversion to CDMS.
In conclusion, our study provides novel insights about CSF contactin-1 and contactin-2 as surrogate markers for axonal domain dysfunction in different MS subtypes, and indicates that these proteins probably reflect novel aspects of the neuro-axonal degenerative mechanism. Therefore, the addition of contactin-1 and contactin-2 to the panel of biomarkers for monitoring axonal damage might be useful.
Supplemental Material
Supplemental material for Contactin-1 and contactin-2 in cerebrospinal fluid as potential biomarkers for axonal domain dysfunction in multiple sclerosis
Supplemental Material for Contactin-1 and contactin-2 in cerebrospinal fluid as potential biomarkers for axonal domain dysfunction in multiple sclerosis by Madhurima Chatterjee, Marleen JA Koel-Simmelink, Inge MW Verberk, Joep Killestein, Hugo Vrenken, Christian Enzinger, Stefan Ropele, Franz Fazekas, Michael Khalil and Charlotte E Teunissen in Multiple Sclerosis Journal—Experimental, Translational and Clinical
Footnotes
Acknowledgements
The authors thank Kees van Uffelen and Harry Twaalfoven for helping with the measurements of contactin-1, contactin-2, NFL and NFH.
Author contribution
MC, MKS and IV performed the experiments. MK provided CSF samples and patient data for the CIS cohort. MC, MK and CT analysed the patient data. MC, MK and CT wrote the paper. MKS, IV, JK, HV, CE, SR and FF provided valuable feedback on the manuscript for intellectual content. MC and CT designed the research study.
Conflict of Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was funded by the European Neuroscience Campus Network, an Erasmus Mundus Joint Doctoral Program (cycle 5/2014/P-04).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.