
Abstract
We describe the case of a man in his 40 s with aggressive multiple sclerosis (MS) who received autologous haematopoietic stem cell transplantation (AHSCT) and subsequently developed probable, if not definite, Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP) and haematological complications. Autoimmune conditions occurring as a side effect of allogenic transplantations are well known in the context of haematological malignancies, but only rarely reported for autologous transplantations. Our case demonstrates that although AHSCT may be effective for suppressing MS inflammatory activity, the profound changes to the immune repertoire may lead to other clinically relevant autoimmune phenomena. A careful benefit-risk evaluation should be conducted in all cases where AHSCT is considered.
Keywords
Case
A previously healthy man aged 42 years was diagnosed with relapsing–remitting multiple sclerosis in 2005, approximately 10 years after his first symptoms. Magnetic resonance imaging (MRI) of the brain and spine showed several bright periventricular and juxtacortical lesions as well as two hyperintense lesions of more than 3 mm but less than two vertebral bodies in length on T2. He was started on interferon-beta and later glatiramer acetate (GA), but discontinued treatment three years after due to side effects and stable conditions. Three months after discontinuing GA, the patient had a clinical relapse supported by MRI findings and was started on natalizumab (NTZ). His Expanded Disability Status Scale (EDSS) score at the time was 3.0 (Figure 1). Despite NTZ, he continued to experience yearly relapses and was treated with steroids with clinical improvement. He was anti-NTZ antibody negative. One year after starting NTZ his EDSS was 4.0, and at three years 5.5. He was anti-John Cunningham (JC) virus antibody positive and treatment with NTZ was discontinued due to risk of progressive multifocal leucoencephalopathy. At this point he had a spastic paraparesis and there were a few new lesions in the spinal cord, including one large lesion in the cervical medulla, but no new lesions on MRI brain. Treatment with fingolimod was suggested before considering referral for autologous haematopoietic stem cell therapy (AHSCT). However, the patient declined and referred himself to Karolinska University Hospital in Stockholm, Sweden, for AHSTC.
EDSS and EDSS equivalent from diagnosis to last clinical contact June 2015. At the time of diagnosis, the patient had reached an EDSS of 2.5. He experienced several clinical relapses, which were supported by MRI findings and his EDSS was 3.0 when natalizumab (NTZ) was introduced four years after diagnosis. Despite treatment with NTZ, he continued to experience clinical relapses and exhibited improvement when treated with steroids. MRI showed only a few medullary lesions on T2 at the time of NTZ discontinuation three years later, and no new lesions on MRI brain. His EDSS was at this time 5.5 due to a spastic paraparesis and this was unchanged as he started BEAM six months later. For the few months post-treatment his EDSS climbed to 7.0, most likely due to side effects of BEAM and repeated infections, though engraftment syndrome cannot be excluded. After some improvement, he once again experienced gait difficulties in September and in January 2014 he was diagnosed with probable CIDP based on neurographic findings and a flaccid paraparesis. EDSS was equivalent to 7.5 at the time of diagnosis. EDSS: Expanded Disability Status Scale; MRI: magnetic resonance imaging; BEAM: carmustine, etoposide, cytarabine and melphalan; CIDP: chronic inflammatory demyelinating polyradiculoneuropathy.
AHSTC was started in November 2012, six months after NTZ was discontinued. He received cyclophosphamide followed by filgrastim for five days. Stem cells were harvested from peripheral blood and a month later he was admitted for carmustine, etoposide, cytarabine, and melphalan (BEAM) conditioning. After treatment he received two days of anti-thymocyte globulin and subsequently experienced seven days of agranulocytosis. During this phase he experienced severe sepsis that was treated with antibiotics. One month after BEAM conditioning he was discharged on ciprofloxacin, trimethoprim, fluconazole, acyclovir and omeprazole. Neutrophil count was 0.61 (1.5–7.0).
At the time of discharge, and in the months following, he had an EDSS corresponding to 7.0. Whether his clinical decline was due to infections, flares of MS associated with treatment or engraftment syndrome is unknown, though MRI shortly after AHSCT did not show any new lesions. He also developed a drug-induced allergic rash and chronic diarrhoea, which was extensively investigated without finding a responsible pathogen. Despite his blood count improving, he experienced repeated minor and major infections. Five months post-AHSCT he restarted the vaccination program and subsequently developed neutropaenia (0.98). Seven months post-AHSCT he had regained limited walking capability and clinical examination confirmed an EDSS 5.5 due to spastic paraparesis. Shortly after, he contracted pulmonary sepsis and an allergic reaction to penicillin. Neutrophils remained low.
Nine months post-AHSCT he once again presented with gradual worsening in gait and fatigue, and clinical examination showed a mixed picture with a spastic gait and weak reflexes. MRI remained unchanged since AHSCT. Nevertheless, he received a course of intravenous (iv) methylprednisolone, but without clinical improvement. One year post-AHSCT he was admitted with lower limb pain and a significant worsening in gait. On clinical examination he had flaccid paraparesis without wasting and areflexia. EDSS was equivalent to 6.5. The patient had symmetrical polyradiculopathy with progression over more than two months. Nerve conduction studies showed sensory-motor polyneuropathy of probable demyelinating type in the lower extremities with pronounced F-response abnormality (Figure 2). The patient had probable, if not definite, chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).
1
He started a five-day course of iv immunoglobulins. However, his neutrophils once again plummeted to 0.4 and the patient remained clinically unchanged. We refrained from giving a second course of immunoglobulins due to neutropaenia and lack of improvement. Brain and cord MRI remained unchanged. Another steroid course offered no improvement. Rituximab was considered but neither the patient nor the neurologists were willing to further blindly manipulate an already weakened immune system. The patient showed no improvement over the next few weeks and nerve conduction velocities remained unchanged. As a last resort, six courses of plasma exchange were tried with no effect. He was wheelchair bound (EDSS equivalent to 7.5).
Summary of neurographic findings at time of diagnosis. The patient had more than two years’ progression of symmetrical polyradiculopathy with areflexia and no wasting. According to the European Federation of Neurological Societies/Peripheral Nerve Society guideline on Management of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP), the patient had probable, if not definite, CIDP. The image on the bottom left shows prolongation of F-wave latency in the left tibial nerve and the image on the bottom right shows amplitude reduction and a partial motor conduction block in the right peroneal nerve.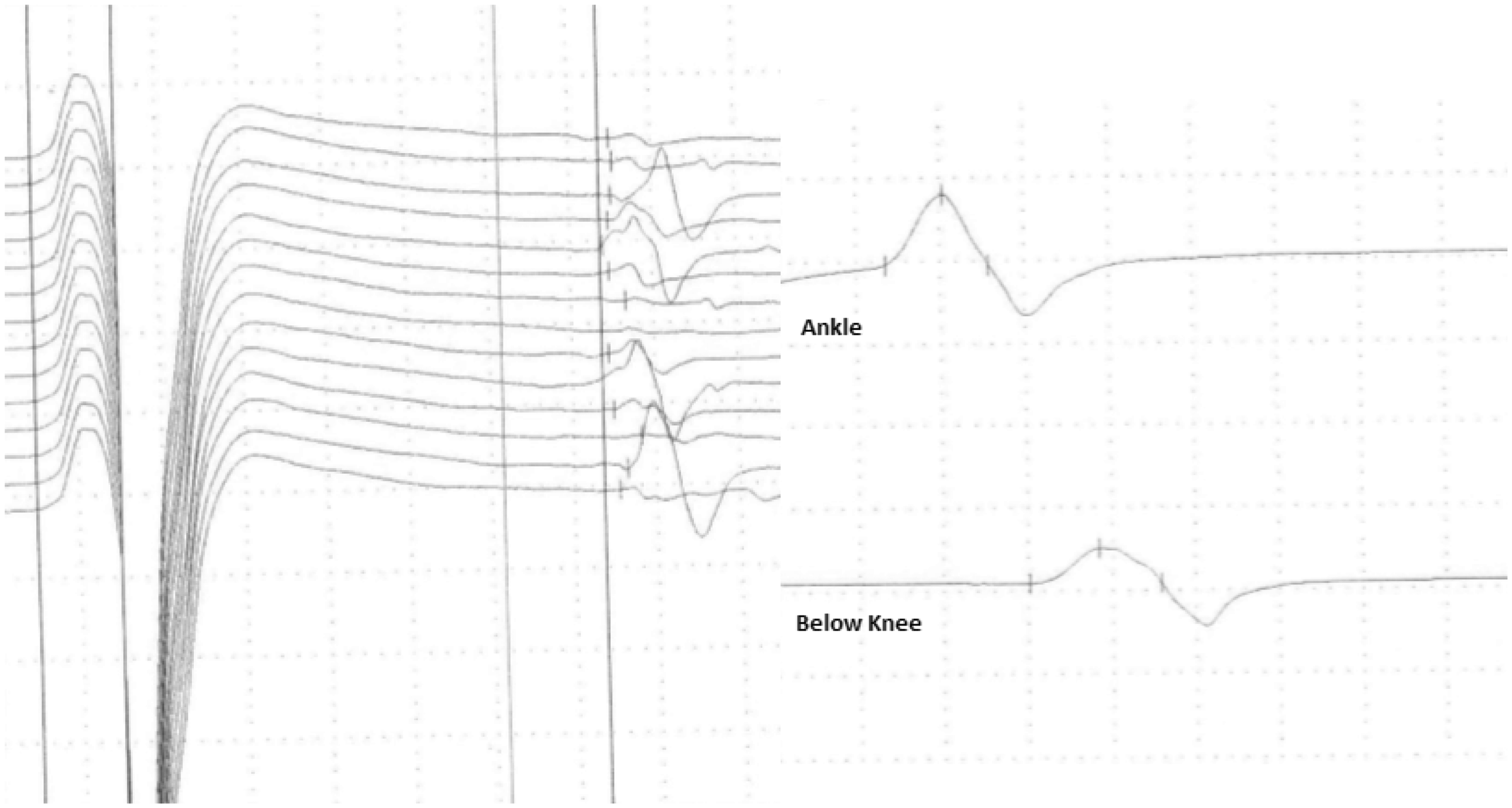
He has since shown gradual improvement and nerve conduction velocities have ameliorated. However, two years post-AHSCT he was once again admitted to hospital with neutropaenic sepsis (neutrophils 0.9) and he was still dependent on two sticks for ambulation, corresponding to EDSS 6.5. We did not find any evidence of other autoimmune diseases. The patient provided written, informed consent to this report.
Discussion
CIDP is a rare, acquired, immune-mediated progressive or relapsing disorder of the peripheral nervous system lasting more than two months, with a strong association to certain bacterial or viral infections but also appearing as a pure autoimmune phenomenon.2,3 Nearly 10% of patients undergoing HSCT for an autoimmune disease have been reported to develop a secondary autoimmune illness within the first three years after HSCT. 4 Inflammatory neuromuscular complications are known to occur secondary to graft versus host disease after allogenic HSCT for haematological malignancies, but then usually as an acute inflammatory demyelinating polyradiculoneuropathy. However, several cases of CIDP after allogenic HSCT have previously been reported.5,6 In contrast, we found only a few cases of CIDP secondary to AHSCT7,8 and no cases secondary to AHSCT for MS in the literature. Autoimmune thyroid disease and cytopaenias are most often described. De novo autoimmunity may develop through loss of central or peripheral immunological tolerance mechanisms. 9 The temporal profile described here is consistent with CIDP occurring as a result of AHSCT, either as a direct consequence or secondary to one of his many septic episodes or the vaccination program.
AHSCT is a promising treatment option for aggressive MS given the evidence of its efficacy and that it alleviates the need for continuous immunosuppression. Still, the procedure is associated with a significant risk of morbidity and a low but not insignificant mortality rate. 10 It is also important to acknowledge that safety outcomes both in the short and long term are less well defined compared to alternative disease-modulatory regimens and that the treatment leads to a permanently changed immune repertoire. This is illustrated by this case report, which also highlights the need for regular and qualified follow-up after AHSCT.
Footnotes
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.