
Abstract
Background
Teriflunomide is a once-daily oral immunomodulator for the treatment of relapsing−remitting MS.
Objective
To evaluate the safety and tolerability of teriflunomide as add-on therapy to a stable dose of glatiramer acetate (GA) in patients with relapsing forms of MS (RMS).
Methods
Phase II, randomized, double-blind, add-on, placebo-controlled study. The primary objective was to assess safety and tolerability; secondary objectives were to evaluate effects of treatment on disease activity assessed by MRI and relapse.
Results
Patients with RMS on GA (N = 123) were randomized 1:1:1 to receive teriflunomide 14 mg (n = 40), 7 mg (n = 42), or placebo (n = 41) for 24 weeks; 96 patients entered the 24-week extension, remaining on original treatment allocation. Teriflunomide was well tolerated over 48 weeks. The frequency of adverse events (AEs) was low across all groups; 5 (12.2%), 3 (7.1%), and 2 (5.0%) patients in the 14 mg, 7 mg, and placebo groups, respectively, discontinued treatment due to AEs. Teriflunomide reduced the number of T1-Gd lesions vs placebo (14 mg: 46.6% relative reduction, p = 0.1931; 7 mg: 64.0%: relative reduction, p = 0.0306).
Conclusions
Teriflunomide added to stable-dose GA had acceptable safety and tolerability, and reduced some MRI markers of disease activity compared with GA alone.
NCT00475865 (core study); NCT00811395 (extension).
Keywords
Introduction
Teriflunomide (AUBAGIO, Genzyme, Cambridge, MA) is a once-daily, oral immunomodulator approved for the treatment of relapsing−remitting MS or relapsing forms of MS (RMS).1,2 In two pivotal phase 3 studies, TEMSO (NCT00134563) and TOWER (NCT00751881), teriflunomide 14 mg significantly reduced the annualized relapse rate (ARR) and the risk of disability progression compared with placebo; teriflunomide 7 mg also significantly reduced ARR compared with placebo.3,4 Teriflunomide monotherapy demonstrated consistent efficacy on MRI parameters in addition to a similar and consistent safety profile with both doses.3,5
In a 48-week phase 2 study, teriflunomide given as add-on therapy to interferon-β (IFN-β) (NCT00489489 [24-week study] and NCT00811395 [24-week extension]) was associated with an acceptable safety and tolerability profile and reductions in MRI disease activity compared with IFN-β alone. 6
In a phase 3 study (TERACLES, NCT01252355), teriflunomide 14 mg added to a stable dose of IFN-β demonstrated an incremental benefit on ARR and a significant reduction (70.8%; p = 0.0061) in the number of gadolinium (Gd)-enhancing T1 lesions/scan compared with IFN-β alone, although early study termination (sponsor decision) limited interpretation. Teriflunomide was well tolerated with no new safety concerns identified compared with other phase 3 monotherapy studies. 7
In this study, we evaluated the safety and tolerability of teriflunomide added to ongoing glatiramer acetate (GA) for up to 48 weeks in patients with RMS.
Patients and methods
Study design
This phase 2, multicenter, randomized, double-blind, placebo-controlled study evaluated the effect of teriflunomide as add-on therapy in patients with RMS receiving a stable dose of GA. After a four-week screening period, patients entered into a 24-week double-blind treatment period; those completing 24 weeks of treatment and still meeting eligibility criteria were given the opportunity to enter a 24-week double-blind extension study, during which patients continued to receive their originally assigned study treatment.
Standard protocol approvals, registrations, and patient consents
This study (NCT00475865 [24-week study] and NCT00811395 [24-week extension]) was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki. The protocol complied with the laws and regulations of the countries where the study was conducted. All patients provided written informed consent prior to study initiation. An independent data monitoring committee evaluated the design, ethical conduct, and safety of the study.
Study population
Patients were recruited from 24 centers in six countries. Patients who met the 2001 McDonald criteria for MS 9 with a relapsing clinical course (with or without progression), aged 18–55 years, Expanded Disability Status Scale (EDSS) score ≤ 5.5, no onset of relapse in the 60 days prior to randomization, and a clinically stable condition for four weeks pre-study were included. All patients were receiving a stable dose of GA for ≥26 weeks prior to screening. Exclusion criteria included: any prior or concomitant use of cladribine, mitoxantrone, azathioprine, cyclophosphamide, cyclosporine, methotrexate, mycophenolate, or natalizumab; prior use of IFN-β, cytokine therapy, intravenous immunoglobulins, or any investigational drug within the preceding 24 weeks. Any patient with persistent or severe infection, and women who were pregnant, breastfeeding, or planning to conceive during the study were also excluded.
Study procedures and randomization
Eligible patients were randomized (1:1:1) to receive once-daily oral teriflunomide 14 mg or 7 mg, or placebo in addition to GA. The ongoing GA regimen was Copaxone® (Baxter Pharmaceutical Solutions LLC for Teva Neuroscience, Inc.) at the recommended dose of 20 mg injected subcutaneously once daily. Randomization was done centrally, by an interactive voice recognition system that generated the randomization list (block size of three) with stratification by country.
Adverse events (AEs) were reported by the patient or noted by the investigator. Laboratory evaluations were performed at screening, baseline, every two weeks during the initial 24-week study, and every six weeks during the 24-week extension phase. MRI scans were conducted at baseline and at Weeks 8, 16, 24, and 48.
Study objectives and assessments
The primary objective was to assess the safety and tolerability of teriflunomide 14 mg and 7 mg versus placebo when added to ongoing treatment with GA in patients with RMS.
Effects on disease activity as measured by MRI parameters and ARR were evaluated as secondary objectives of this study.
The main MRI parameters assessed were the total number of Gd-enhancing T1 (T1-Gd) lesions and total volume of T1-Gd lesions per scan; other parameters included total volume of T2- and T1-hypointense lesion components, post-Gd T1-hypointense lesion (black holes) volume, T2 lesion volume, average number of unique active lesions per scan (Gd-enhanced lesions on T1-weighted or non-enhanced new or enlarged T2 lesions), atrophy (normalized brain volume), white and gray matter volume, and Z4 composite score (sum of individual Z scores for total lesion volume, T1-Gd-enhancing lesion volume, T1-hypointense lesion volume, and normalized volume of cerebrospinal fluid). MRI results were processed and analyzed at the MRI Analysis Center, University of Texas Health Science Center, Houston, Texas, USA.
A relapse was defined as the appearance of new clinical signs or symptoms or clinical worsening of a previous sign or symptom (that had been stable for four weeks or more) that persisted for a minimum of 24 hours in the absence of fever. Each relapse was confirmed by the treating neurologist based on the objective assessment by the examining neurologist (blinded to treatment allocation). The examining neurologist had to document either: a 1-point increase in two or more functional system (FS) functions or a 2-point increase in one or more FS function (excluding bowel/bladder and cerebral); or an increase of ≥0.5 points in the EDSS score (or an increase of ≥1 if the EDSS score was 0) from the previous clinically stable assessment.
Statistical analysis
The safety population was defined as all randomized patients exposed to study medication; analyses were conducted according to the treatment received and evaluation was based on review of individual values and descriptive statistics. Efficacy analyses were performed on the modified intent-to-treat population defined as all randomized patients who took at least one dose of the double-blind study medication; analyses were conducted according to the group to which patients were randomized. The sample size estimation was based on the primary objective of estimating the safety and tolerability of a 14 mg and a 7 mg dose of teriflunomide, compared with placebo.
The number of T1-Gd lesions per MRI scan was compared between treatment groups using a Poisson model with robust variance estimation, with treatment group, region, and baseline number of T1-Gd lesions as covariates; log-transformed number of scans was included in the model as an offset variable. The total volume of T1-Gd lesions per MRI scan was analyzed using a permutation test; the Monte Carlo approach was used to select a random subset of the total number of permutations of the total volume of T1-Gd lesions per MRI scan (holding independent variables fixed [i.e. baseline, region, and treatment group]). Each permutation sample was analyzed using the analysis of covariance model, adjusting for baseline volume of T1-Gd lesions, treatment, and region. Change from baseline in total lesion volume was analyzed using a mixed-effect model with repeated-measures approach on cubic root transformed volume data.
ARR was analyzed using a Poisson regression model with robust variance estimation, which included the total number of confirmed relapses with onset between randomization date and last dose date as response variable, with treatment group and region as covariates; log-transformed standardized treatment duration was included in the model as an offset variable.
Results
Study population
Between April 2007 and December 2008, 148 patients were screened and 123 were randomized (teriflunomide 14 mg, 40; teriflunomide 7 mg, 42; placebo, 41) at 24 centers in Austria, Canada, Germany, Italy, UK, and USA; 96 patients entered the 24-week extension phase (teriflunomide 14 mg, 28; teriflunomide 7 mg, 30; placebo, 38) (Figure 1).
Study disposition
Baseline demographics and clinical variables (safety population).

EDSS = Expanded Disability Status Scale; Gd = gadolinium; mo = months; SD = standard deviation; y = years.
Safety and tolerability
Overview of treatment-emergent adverse events (safety population).

Treatment-emergent adverse events by Medical Dictionary for Regulatory Activities (MedDRA) preferred term ≥10% in any group at 48 weeks, ranked by decreasing order in the teriflunomide 14 mg + GA group.
MedDRA preferred term, alopecia.
ALT = alanine aminotransferase; GA = glatiramer acetate; ULN = upper limit of normal.
Serious adverse events
Thirteen treated patients experienced a total of 19 SAEs by Week 48 of the study (Appendix Table 1); eight of these events occurred in seven patients during the initial 24-week study. Two patients in the 7 mg group experienced an SAE of alanine aminotransferase (ALT) increase, and although the increases were mild and asymptomatic, they were classified by the investigator as medically important. One experienced ALT < 3 × upper limit of normal (ULN) considered treatment-related (normalized two weeks later) and the other reported intermittent ALT 2 × ULN (normalized more than four months later). Serious infections were reported in one (2.4%) patient in the 7 mg group and two (5.0%) patients in the placebo group; no serious infections occurred in patients on 14 mg. One patient in the 7 mg group with a history of excessive smoking and resting dyspnea was diagnosed 71 days after starting treatment with suspected interstitial lung disease (ILD). The diagnosis was never confirmed by bronchoalveolar lavage or lung biopsy, and the patient recovered.
Discontinuations due to AEs
Of the 10 patients who discontinued, seven discontinued during the core study. Reasons for treatment discontinuation included: 14 mg, hair loss (n = 2), moderate fibromyalgia (n = 1), moderate diarrhea (n = 1), seborrheic dermatitis (n = 1); 7 mg, rash (n = 1), dyspnea (n = 1), suspected ILD (n = 1); placebo, herpes zoster (n = 1), headache (n = 1). The case of diarrhea and both instances of hair loss resolved after treatment discontinuation; the herpes zoster case and suspected ILD, discussed above, were classified as serious.
AEs of special interest
Gastrointestinal AEs, including nausea and diarrhea, were more frequently reported in patients treated with teriflunomide compared with placebo (Table 2). All events of nausea were mild-to-moderate in intensity and none led to treatment discontinuation. Diarrhea was mainly mild-to-moderate in intensity (severe in one patient in each teriflunomide group) and led to permanent study discontinuation in one patient (14 mg group; patient recovered after two days).
Infections and hematologic disorders occurred at a higher frequency in the placebo group (67.5%) compared with the 14 mg and 7 mg groups (51.2% and 52.4%, respectively) during the core and extension phases of this study; the majority of first onset of events occurred during the core phase of the study. The proportion of patients with decreased neutrophil counts (<1500 cells/µL) was slightly higher in teriflunomide-treated patients (14 mg, [n = 4] 9.8%; 7 mg, [n = 3] 7.1%) compared with placebo (n = 1, 2.5%); however, these events were not considered serious, and did not lead to treatment discontinuation. There were no events of neutrophil counts <1000 cells/µL.
The proportion of patients with hair loss was higher in the 14 mg group (17.1%) compared with the other groups (7 mg [11.9%]; placebo [2.5%]); all instances of hair loss were mild or moderate in intensity. All cases occurred in the initial 24-week phase; approximately half resolved within four months.
Laboratory assessments
The proportion of patients with increased ALT was low and similar across treatment groups. One patient in the placebo group had ALT increase >15 × ULN and aspartate transaminase (AST) >8 × ULN, 10 weeks after the cholestyramine washout period following the core study. One patient in the 14 mg group had two transient episodes of ALT > 5 ×ULN, once during the core study, and the other during the extension.
No cases meeting Hy’s Law criteria (ALT > 3 × ULN and total bilirubin > 2 × ULN) were reported. No patients discontinued treatment because of ALT increase (patients with ALT > 3 × ULN confirmed within 48 hours were required to permanently discontinue study treatment).
One patient in the 14 mg group presented with serum amylase >2 × ULN. No pancreatic abnormalities were observed (CT or MRI). Asymptomatic lipase increases >2 × ULN were observed in four patients (three in the 14 mg group, one in the placebo group) at various time points during the core and extension studies; all participated in the extension study and were without abdominal ultrasound abnormalities. None of the patients discontinued study treatment because of increased lipase and all recovered within one month of each separate elevation.
Efficacy of adjunctive treatment
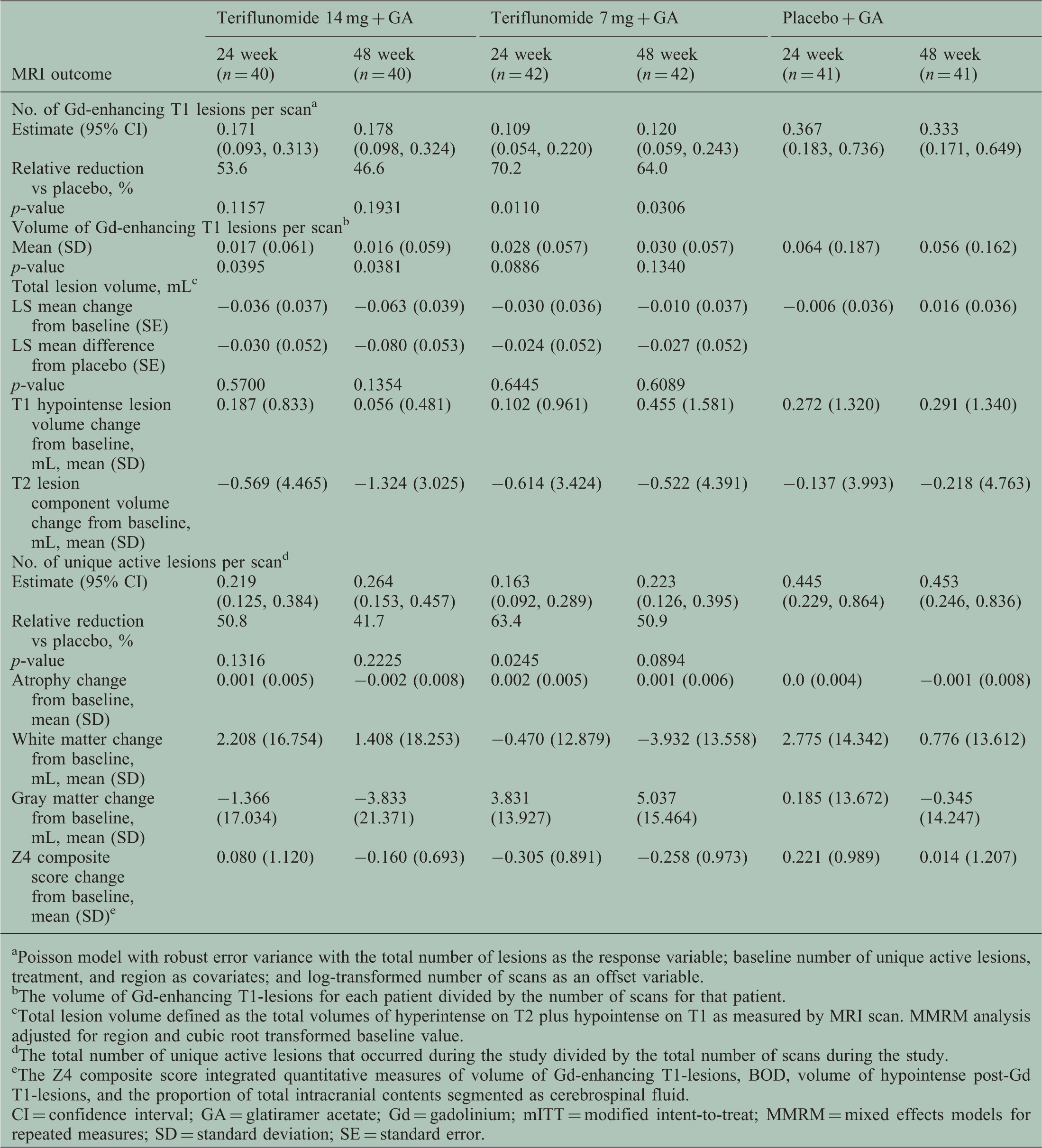
A numerical reduction was observed in the number of T1-Gd lesions per scan in the 14 mg group compared with placebo at 24 and 48 weeks (53.6% and 46.6% reductions, respectively), without achieving statistical significance. Teriflunomide 7 mg significantly reduced the number of T1-Gd lesions per scan at both time points (70.2% [p = 0.0110] and 64.0% [p = 0.0306] reductions, respectively, compared with placebo). Total T1-Gd volume per scan was significantly reduced in the 14 mg group compared with placebo at both 24 and 48 weeks (73.0% [p = 0.0395] and 73.1% [p = 0.0381] reduction, respectively). A numerical reduction in T1-Gd volume was observed in the 7 mg group compared with placebo at both time points (Figure 2). Other MRI outcomes are reported in Table 3.
MRI outcomes of teriflunomide as an adjunct to glatiramer acetate (mITT population). (a) Number of Gd-enhancing T1 (T1-Gd) lesions per scan at 24 and 48 weeks (the total number of T1-Gd lesions that occurred during the study divided by the total number of scans during the study, adjusted for baseline number of T1-Gd lesions, treatment, and region using a Poisson model with robust variance estimation). Error bars represent 95% confidence intervals. (b) Volume of T1-Gd lesions per scan at 24 and 48 weeks. The total volume of lesions that occurred during the study divided by the total number of scans during the study. MRI outcomes (mITT population). Poisson model with robust error variance with the total number of lesions as the response variable; baseline number of unique active lesions, treatment, and region as covariates; and log-transformed number of scans as an offset variable. The volume of Gd-enhancing T1-lesions for each patient divided by the number of scans for that patient. Total lesion volume defined as the total volumes of hyperintense on T2 plus hypointense on T1 as measured by MRI scan. MMRM analysis adjusted for region and cubic root transformed baseline value. The total number of unique active lesions that occurred during the study divided by the total number of scans during the study. The Z4 composite score integrated quantitative measures of volume of Gd-enhancing T1-lesions, BOD, volume of hypointense post-Gd T1-lesions, and the proportion of total intracranial contents segmented as cerebrospinal fluid. CI = confidence interval; GA = glatiramer acetate; Gd = gadolinium; mITT = modified intent-to-treat; MMRM = mixed effects models for repeated measures; SD = standard deviation; SE = standard error.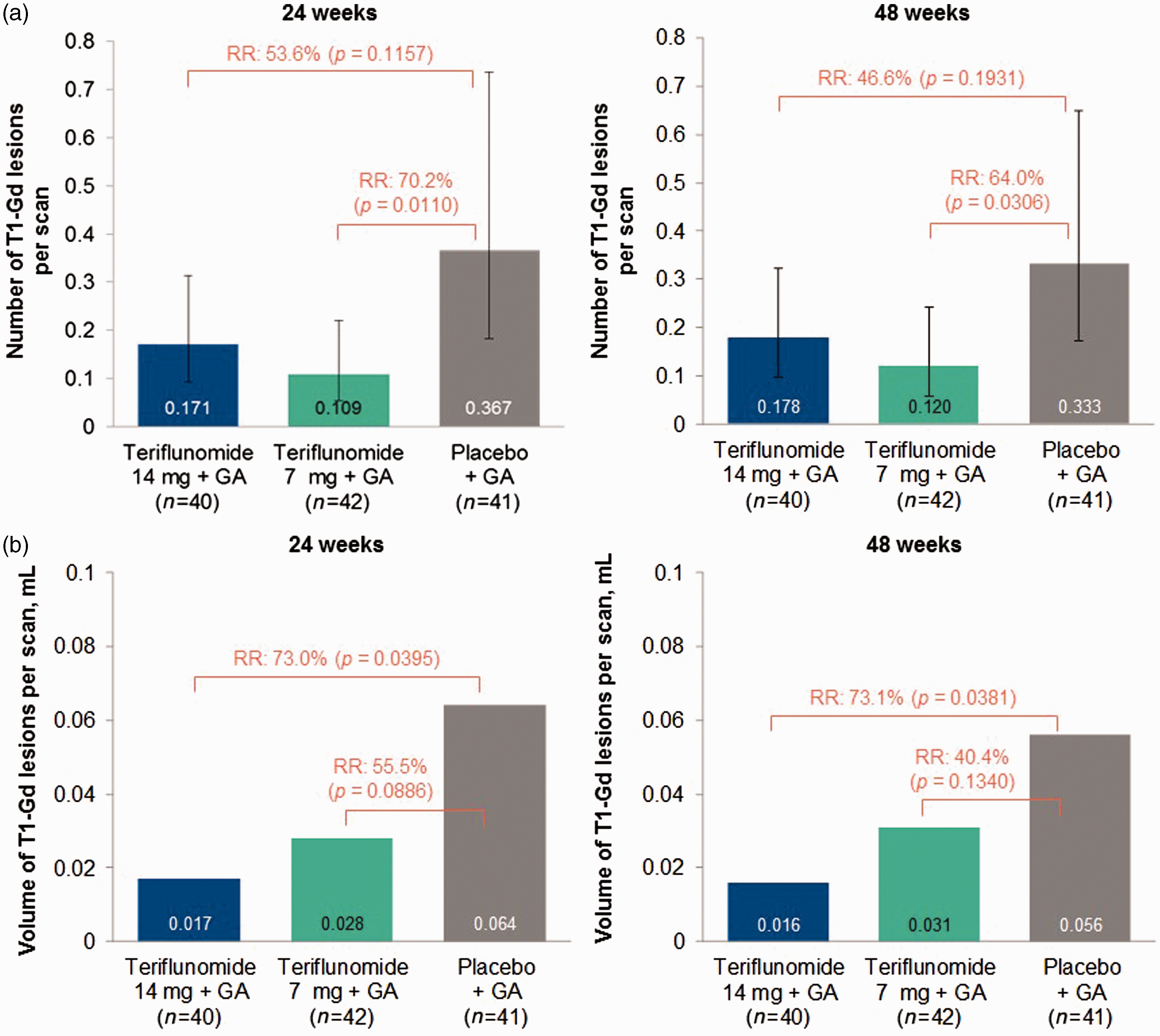
Adjusted ARRs were 0.497, 0.262, and 0.420, in the 14 mg, 7 mg, and placebo groups, respectively.
Discussion
This study was designed to assess the safety of teriflunomide 14 mg or 7 mg as add-on therapy to stable-dose GA in patients with RMS. Both doses of teriflunomide added to GA demonstrated acceptable safety and tolerability compared with placebo plus GA during the core study and extension phase. After 48 weeks of treatment, the proportion of patients reporting at least one AE was similar among all treatment groups. Few patients experienced SAEs or AEs leading to treatment discontinuation. Only one patient in each of the 14 mg and placebo groups had ALT increases >5 × ULN, and no cases meeting Hy’s Law criteria were reported. Although the proportion of patients with AEs related to hematologic parameters (neutrophils <1500 cells/µL) was higher in teriflunomide-treated patients, none of the events led to treatment discontinuation. The nature and incidence of AEs in this study are comparable to findings from teriflunomide monotherapy studies, TEMSO and TOWER,3,4 as well as safety observations from a study of teriflunomide as an add-on therapy to IFN-β. 6 Our data support the safety of teriflunomide when added to ongoing GA therapy and, by extension, as a sequential treatment for patients previously treated with GA.
This study was not sufficiently powered to assess the efficacy of teriflunomide as an add-on therapy to GA; the lack of a teriflunomide-only arm, and the greater baseline MRI disease activity in the 7 mg group, further limit interpretation of efficacy outcomes. Nevertheless, we observed a general pattern of lower MRI activity in the teriflunomide groups compared with placebo, indicative of a lack of a negative drug–drug interaction between teriflunomide and GA. We also noted a significant reduction in the number of T1-Gd lesions in the 7 mg group and a reduction in T1-Gd lesion volume in the 14 mg group at 24 and 48 weeks. There were no significant differences between treatment groups with regard to clinical measures.
Combination therapy is widely used in many medical conditions, including rheumatoid arthritis, hypertension, and diabetes mellitus, and is generally accepted to provide improved disease management; however, currently, all licensed disease-modifying therapies (DMTs) for MS are approved as monotherapy and American Academy of Neurology Guidelines do not offer recommendations for combination therapy in MS. 11 Despite numerous studies of combination therapy with DMTs,10–12 its role in MS management remains poorly defined, although safety profiles were generally acceptable.13,14
Patients and their treating physicians may consider switching DMTs for reasons such as suboptimal response, treatment fatigue due to route of administration, poor tolerability, poor adherence, lifestyle choices, or financial reasons.15–17 In the TEMSO and TOWER trials, 28 − 34% of patients in each teriflunomide group used DMTs (IFNβ-1a, IFNβ-1b, and GA) in the two years before study entry; however, study protocols required no use of GA or IFN within three (TOWER) or four (TEMSO) months prior to randomization, and prior use of natalizumab was prohibited. No new or additional safety concerns were identified in patients previously treated with DMTs and subsequent treatment with teriflunomide.3,4
It is interesting to compare the MRI findings from this study with a similar study evaluating the safety of teriflunomide in combination with IFN-β. 6 That study showed evidence of an additive effect of teriflunomide and IFN-β compared with IFN-β alone on MRI outcomes and a trend for superiority of the combination regarding relapses. There may be a number of possible explanations for this difference; for example, it may relate to differences in the interactions of individual agents, or differences in the study populations. Importantly, comparisons across studies are associated with many other limitations. The positive safety observations from this study provide assurance for the safety of overlapping usage for short duration, and suggest no concern about immediate switching between GA and teriflunomide therapies.
Footnotes
Acknowledgements
This study was funded by Genzyme, a Sanofi company. This manuscript was reviewed by Stephanie Jurgensen, Deborah Dukovic and Karthinathan Thangavelu, of Genzyme. Editorial support was provided by Scott Chambers, Fishawack Communications Inc., also funded by Genzyme. Essential contributing members of the MRI Analysis Center (Houston, TX, USA) included Professor Ponnada A. Narayana, Dr Flavia Nelson, Dr Sushmita Datta, Dr Irina Vainrub, Brandi Carrico, Kelly Ton, Rimma Brokhin, and Lucille Lambert. Huiling Li, an employee of Sanofi, was responsible for statistical analyses.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Genzyme, a Sanofi company.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Professor Freedman received consulting fees from Bayer HealthCare, Biogen Idec, Chugai, EMD Canada, Genzyme, Novartis, Sanofi, and Teva Canada Innovation; served on Advisory Boards or Boards of Directors for Bayer HealthCare, Biogen Idec, Hoffman La-Roche, Merck Serono, Novartis, Opexa, and Sanofi; and speaker’s bureau for Genzyme. Professor Wolinsky received consulting fees from AbbVie, Actelion, Alkermes, Athersys, EMD Serono, Forward Pharma, Genentech, Genzyme, Novartis, Roche, Teva, Teva Neuroscience, to-BBB, and XenoPort; support for contracted research from Genzyme, the National Institutes of Health and the National Multiple Sclerosis Society through the University of Texas Health Science Center at Houston; and royalty payments through the University of Texas Health Science Center at Houston for monoclonal antibodies out-licensed to Chemicon. Dr Truffinet is an employee of Genzyme, a Sanofi company, with ownership interests. Professor Comi received consulting fees from Almirall, Bayer, Chugai, Excemed, Genzyme, Merck Serono, Novartis, Receptos, Sanofi, Serono Symposia International Foundation (SSIF), and Teva; fees for non-CME services from Almirall, Bayer, Biogen, Excemed, Genzyme, Merck Serono, Novartis, Receptos, Sanofi, SSIF, and Teva. Professor Kappos’ institution has received in the last three years and used exclusively for research support, steering committee, advisory board and consultancy fees from Actelion, Addex, Bayer HealthCare, Biogen Idec, Biotica, Genzyme, Lilly, Merck, Mitsubishi, Novartis, Ono Pharma, Pfizer, Receptos, sanofi-aventis, Santhera, Siemens, Teva, UCB, and XenoPort; speaker fees from Bayer HealthCare, Biogen Idec, Merck, Novartis, sanofi-aventis, and Teva; support for educational activities from Bayer HealthCare, Biogen, CSL Behring, Genzyme, Merck, Novartis, Sanofi, and Teva, royalties from Neurostatus Systems GmbH; and grants from Bayer HealthCare, Biogen Idec, European Union, Merck, Novartis, Roche Research Foundation, Swiss MS Society, and the Swiss National Research Foundation. Dr Miller has received consulting fees from Accordant Health Services, Acorda Therapeutics, Alkermes, Biogen Idec, EMD Serono, Genentech, Genzyme, GSK, Mallinckrodt Pharmaceuticals (Questcor) Roche and Teva; and contracted research fees from Biogen Idec, Genentech, Novartis, Questcor, Roche, and Sanofi. Dr Olsson has received consulting fees from Biogen Idec, Genzyme, and Novartis; speakers’ fees from Teva; and contract research support from Biogen Idec, Genzyme, and Novartis. Dr Benamor is an employee of Genzyme, a Sanofi company. Dr Chambers is an employee of Fishawack Communications Ltd. Dr O’Connor has received consulting fees from Actelion, Bayer, Biogen Idec, BioMS, Cognosci, Daiichi Sankyo, EMD Serono, Genentech, Genmab, Novartis, Roche, Sanofi, and Teva; and contracted research fees from Actelion, Bayer, Biogen Idec, BioMS, Cognosci, Daiichi Sankyo, EMD Serono, Genentech, Genmab, Novartis, Roche, Sanofi, and Teva.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.