
Abstract
Background:
Severe acute kidney injury (AKI) in the intensive care unit (ICU) is complicated by fluid accumulation, making fluid removal a central component of kidney replacement therapy (KRT). However, the optimal fluid management strategy in patients receiving KRT remains unknown, and practice varies widely.
Objective:
To assess the feasibility of conducting a multicentre randomized controlled trial comparing a protocol-based fluid-removal strategy with usual care in critically ill adult patients receiving KRT. The primary objective is to determine whether the intervention results in a difference in cumulative fluid balance from randomization to day five.
Design:
Open-label, multicentre, pilot randomized controlled trial.
Setting:
Centers in Canada, the United States, and Australia.
Patients:
We are enrolling 150 adults admitted to the ICU with AKI who have been receiving KRT for ≤48 hours or who are anticipated to commence KRT within the next 12 hours.
Measurements:
The primary outcome is the difference in cumulative fluid balance (mL) between treatment arms from randomization (day zero) to the end of day five. Secondary outcomes include feasibility metrics, patient outcomes, resource use, safety outcomes, and process measures.
Methods:
Participants are randomized 1:1 to receive either protocol-based fluid management or usual care. The intervention consists of a prescription template updated at least once daily by the attending care team, specifying a 24-hour fluid balance target, a prescription for fluid removal using KRT, and daily re-evaluation of fluid intake. The intervention is continued until day five post-randomization, KRT discontinuation due to kidney recovery, or ICU discharge.
Limitations:
The application of the intervention relies on the clinical judgment of the attending care team, which may affect the fidelity of the intervention. Usual care may differ between institutions, which may lead to variability. The treatment teams are unblinded, however the statistician will be blinded to group allocation.
Conclusions:
The Probe-Fluid pilot trial will provide important groundwork toward a future definitive multicentre randomized controlled trial comparing a protocol-based fluid management strategy with usual care in critically ill patients receiving KRT.
Introduction
Severe acute kidney injury (AKI) in the intensive care unit (ICU) is almost uniformly complicated by fluid accumulation, making fluid removal a central component of the kidney replacement therapy (KRT) prescription. 1 Multi-organ congestion resulting from fluid accumulation is believed to mediate adverse outcomes, including pulmonary congestion, systemic inflammation, and impaired hepatic function.2-15 Furthermore, positive cumulative fluid balance at the time of KRT initiation is associated with higher mortality and less kidney recovery.16-18 Accordingly, rapid treatment of fluid overload and prevention of organ congestion may help improve patient outcomes. On the other hand, the risk of hemodynamic instability is increased in acutely ill patients undergoing KRT due to alterations in the compensatory physiological mechanisms in response to fluid removal. 19 Hemodynamic Instability Related to Renal Replacement Therapy occurs in about 10% to 30% of KRT sessions in the ICU,20,21 exposing patients to potential iatrogenic organ hypoperfusion. Thus, balancing the benefits and harms of fluid removal remains a major clinical challenge.
The optimal fluid management strategy is currently unknown. Recent international surveys of intensivists and nephrologists highlighted significant variability in fluid management practices for critically ill patients receiving KRT. 22 This heterogeneity underscores the need to address important knowledge gaps through the conduct of randomized clinical trials (RCT) on this topic, which was identified as a research priority during the 17th Acute Disease Quality Initiative (ADQI XVII) conference. 23 Trials that have been completed to date reported significant challenges in patient recruitment, protocol adherence, and provider acceptability. 24 Therefore, it is important to demonstrate the feasibility of conducting a multicentre RCT using a clinical decision support tool that could realistically scale up to a definitive trial evaluating patient-centered outcomes.
We have launched a pilot multicentre RCT comparing a protocol-based fluid management strategy with usual care in critically ill patients receiving KRT. The protocol is designed to promote the achievement of a neutral or negative daily fluid balance early in the course of KRT treatment by both preventing and treating fluid accumulation. It provides a clinical decision support for prescribing fluid management on KRT while allowing the attending care team to flexibly modify treatment targets according to their clinical evaluation. Herein, we describe the Probe-Fluid pilot trial protocol.
Methods
Study Design and Setting
Proactive prescription-based fluid management versus usual care in critically ill patients on kidney replacement therapy (Probe-Fluid) is an open-label, multicentre, pilot RCT comparing a prescription-based fluid management strategy with usual care in critically ill adult patients with AKI for whom KRT has been, or will shortly be, initiated. The trial is active in eight centers in Canada, the United States, and Australia. The protocol is summarized according to the guidelines set out in the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 Statement which is provided in Supplemental Material. 24
Population and Eligibility
The trial is recruiting adults (age ≥18 years) admitted to the ICU with AKI during the current hospitalization as defined by the KDIGO (Kidney Disease: Improving Global Outcomes) criteria 1 and who are planned to start KRT within the following 12 hours or have been receiving KRT for ≤48 hours. Detailed inclusion and exclusion criteria are outlined in Table 1. Patients who are initially ineligible due to potentially reversible criteria may be re-screened for study enrolment. The trial allows the application of any consent mechanism that has been approved by the local ethics boards and regulatory authorities in that jurisdiction.
Inclusion and Exclusion Criteria for the Probe-Fluid Trial.

Note. ICU = intensive care unit; AKI = acute kidney injury; KDIGO = Kidney Disease: Improving Global Outcomes; KRT = kidney replacement therapy.
Randomization
Following verification of eligibility, randomization is performed by research personnel using a computer-generated sequence with block randomization of variable random sizes,2-6 stratified by site, via a web-based central randomization system (Research Electronic Data Capture [REDCap]). Participants are randomized in a 1:1 ratio to one of two treatment strategies: protocol-based fluid management versus usual care. The randomization methodology and the generated allocation sequence are concealed from the investigators and participants.
Intervention Arm
The intervention is comprised of three components that are initiated immediately after randomization. It consists of a clinical decision support tool consisting of a prescription template with directions on monitoring and safety thresholds for net fluid-removal rate updated by the attending care team at least once daily, before noon. The template is summarized in Figure 1 and presented in detail in the Supplemental Material.

Summary of components of the protocol-based fluid management prescription.
The first component of the intervention is to define the 24-hour fluid balance target, either by:
Option 1 (default): Aiming for a negative fluid balance of 2% to 3% body weight (e.g., 1.4-2.1 L in a 70 kg participant);
Option 2 (alternative): Aiming to avoid fluid accumulation by targeting a neutral fluid balance within 0.5% of body weight variation (e.g., −350 to +350 mL in a 70-kg participant).
The second component is to pre-specify a prescription for fluid removal using KRT as follows:
For patients on continuous KRT, re-evaluation of fluid balance targets is performed by a member of the healthcare team at least every eight hours, and more frequently as needed, in order to re-adjust the net fluid-removal rate and achieve the desired fluid balance by the end of the 24-hour period. Safety thresholds, including arterial pressure, vasopressor support, or any other clinical parameters deemed to be of importance, are pre-specified by the attending care team. Reaching any of these thresholds will prompt a cessation of fluid removal by the nurse in charge of KRT management, followed by a re-evaluation of fluid-removal goals and safety thresholds by the attending care team and an updated target for the remainder of the 24-hour period prescribed by the attending physician.
The use of intermittent KRT is allowed, provided that the net negative fluid-removal goal can be realistically achieved using a maximum net fluid-removal rate of 8 mL/kg/h, since higher targets are frequently unattainable in practice. 25 An increase in the length of intermittent KRT, an increase in the frequency of KRT sessions, or a transition to continuous KRT are advised if this is not possible. Safety thresholds prompting a reduction in fluid-removal rate during KRT sessions are also provided by the attending care team to the dialysis nurse.
The third component is to prompt a daily re-evaluation of fluid intake by the attending care team, including the ICU pharmacist, if available. Specific instructions provided include: (1) Maintenance IV fluids should be avoided. (2) Enteral nutrition, enteral tube irrigation, and IV medications should be given with the least possible amount of fluid. (3) Periods of intravascular expansion using crystalloids, colloids, and blood products are permitted but should be prescribed as a total amount over a time period (e.g., 1 L of Ringer Lactate over four hours) except in special circumstances in which ongoing fluid losses require continued administration.
The intervention is continued until day five post-randomization, KRT discontinuation due to kidney recovery, or ICU discharge. After this, fluid balance management is left to the discretion of the attending care team.
Control Arm
Fluid balance management is not protocolized and is prescribed and adjusted by the attending care team without any specific guidance. The use of the documents provided for the intervention group is not permitted in the control arm.
Concomitant Treatments and Co-Interventions
All other aspects of the KRT prescription are delivered at the discretion of the attending clinicians. This includes all dialysis parameters, including type of treatment, dose (total effluent rate or solute clearance), dialysate temperature, composition, and anticoagulation during KRT. The use of diuretics is permitted in both arms of the trial.
While the protocolized fluid management strategy is updated at least once a day in the intervention group, there is flexibility to make changes at anytime in response to any abrupt changes in the patient’s status. Volume expansion using fluid boluses is allowed if perceived to be clinically indicated by the attending care team, but the fluid management prescription should be updated to reflect revised fluid-removal goals when this occurs.
Definition of Net Fluid Balance
For the purpose of the trial, net fluid balance is defined as the difference between the sum of all quantifiable fluid intake (oral intake, enteral feeding, intravenous intake) and all quantifiable fluid output, including urine output, drainage losses, and net ultrafiltration performed using KRT.
Outcomes
The primary outcome of this study is the difference in cumulative fluid balance (mL) between both treatment groups from randomization through the end of day five. Exploratory analyses will examine differences in cumulative fluid balance (mL) on days one, two, three, and four. Secondary outcomes include feasibility metrics, patient outcomes, resource use, safety outcomes, and process measures (Table 2).
Study Secondary Outcomes.

Note. KRT = kidney replacement therapy; ICU = intensive care unit; SOFA = sequential organ failure assessment.
Adverse Events
All deaths that occur during the five-day follow-up period are reviewed by the local investigators in coordination with the attending clinicians to determine whether these may be linked to participation in the trial. As this trial is recruiting in a population that is already in a life-threatening situation, it is expected that many participants will experience adverse events (AEs). 26 For this study, a reportable AE is defined as an event that is at least possibly related to study procedures or KRT. All reportable AEs are documented and followed up within seven days if fatal or life-threatening. An AE is designated as severe if it meets one of the following criteria: it is fatal, felt to be life-threatening, requires the prolongation of an existing hospitalization, or results in significant disability or incapacity. We defined a list of AEs of special interest potentially related to fluid removal for which reporting is mandatory. These include stroke, myocardial infarction, mesenteric ischemia, digital ischemia, arrhythmias, and cardiac arrest.
Data Collection
Data is collected by trained research personnel using electronic case report forms (Table 3). Baseline data include age, sex, comorbidities, reason for ICU admission, time since ICU admission, time since initiation of KRT and initial KRT modality, cumulative fluid balance from ICU admission to the time of KRT initiation and at enrolment, urine output in the 24 hours prior to enrolment, vasopressor/inotropic support (vasoactive-inotropic score [VIS]27-30), respiratory support mode, PaO2/FiO2, and positive end-expiratory pressure (if any) at enrolment, routine laboratory measurements including platelet count and serum bilirubin, and Sequential Organ Failure Assessment (SOFA) score at enrolment.
Data Collection Schedule.
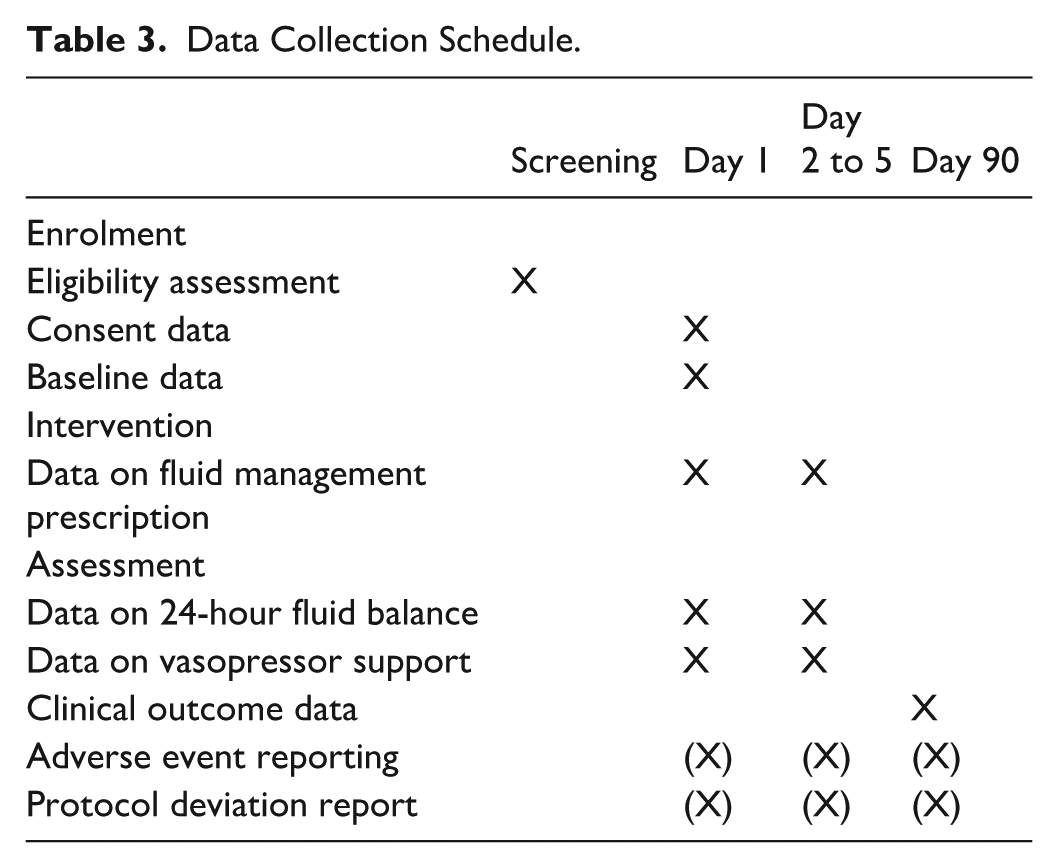
Daily data during the treatment period (up to day five) include KRT modality and duration, detailed daily cumulative fluid balance, measured body weight (when possible), information about the severity of critical illness including SOFA score (total and non-renal component), maximal vasopressor support (VIS), respiratory and oxygen support therapies, and details of fluid-removal prescription in the intervention arm.
Data is obtained at 90 days by chart review and through direct contact with the patient if necessary. This includes the number of days in the ICU and the hospital since enrolment up to 90 days, the number of days receiving intravenous vasopressor support, mechanical ventilation, and KRT since enrolment to 28 days, use of KRT at 90 days, and mortality at 90 days.
Sample Size Considerations
The minimally clinically important difference in cumulative fluid balance in the span of 5 days on KRT is unclear. Based on various sources18,31-35 and preliminary analysis of fluid balance data from the STARRT-AKI trial, 36 the standard deviation (SD) of the cumulative fluid balance at day five after KRT initiation is not precisely known but is estimated to vary from 3000 mL to 6000 mL. With a significance level (α) of 0.05 using a two-sided two-sample equal-variance t-test, group sample sizes of 71 achieved 80% power to reject the null hypothesis of equal means when the population mean difference is 1420 mL (SD of 3000 mL for both groups), or when the population mean difference is 2841 mL (SD 6000 mL). To account for a potential loss to follow-up of 5%, we inflated the planned sample size from 142 to 150 participants.
Statistical Analysis
Statistical analysis will be performed by a statistician blinded to group attribution. All analyses will follow the intention to treat principle (i.e. all patients will be analyzed according to the group to which they were assigned). A P-value of <.05 will be considered as significant. Data will only be analyzed at the end of the trial when patient recruitment and follow-up have concluded. The following statistical analysis method was determined by the investigators without access to the ongoing study data.
The primary endpoint is the cumulative fluid balance (mL) at the end of day five. Missing values are expected due to death or ICU discharge. Therefore, the treatment effect will be estimated using a mixed-effects model for repeated measures (MMRM), the approach recommended in the EMA missing-data guideline and the ICH E9(R1) addendum for longitudinal continuous outcomes 1. Fixed effects will include treatment group, study day (categorical, days one to five), the treatment-by-day interaction, baseline cumulative fluid balance, urine output in the 24 hours prior to enrolment, age, and baseline SOFA score.37,38 Within-patient errors will be modeled with an unstructured variance–covariance matrix. Parameters will be estimated using restricted maximum likelihood (REML). We will apply the Kenward–Roger correction to adjust standard errors and denominator degrees of freedom.
Pre-specified sensitivity analyses of the primary outcome will be performed using multiple imputation (with random treatment allocation, death by day five, ICU discharge by day five, baseline/enrolment prognostic variables, and daily cumulative fluid balance up to day five as variables in the imputation model) and by excluding cases with missing values (complete-case analysis). Exploratory analyses will consider the difference in cumulative fluid balance at one, two, three, and four days. We will also perform the analysis while excluding enteral feeding from the cumulative fluid balance calculation. The statistical analysis method for secondary outcomes is presented in Table 4. No statistical adjustment for multiplicity is planned due to the pilot nature of this trial.
Statistical Analysis Plan for Secondary Outcomes.
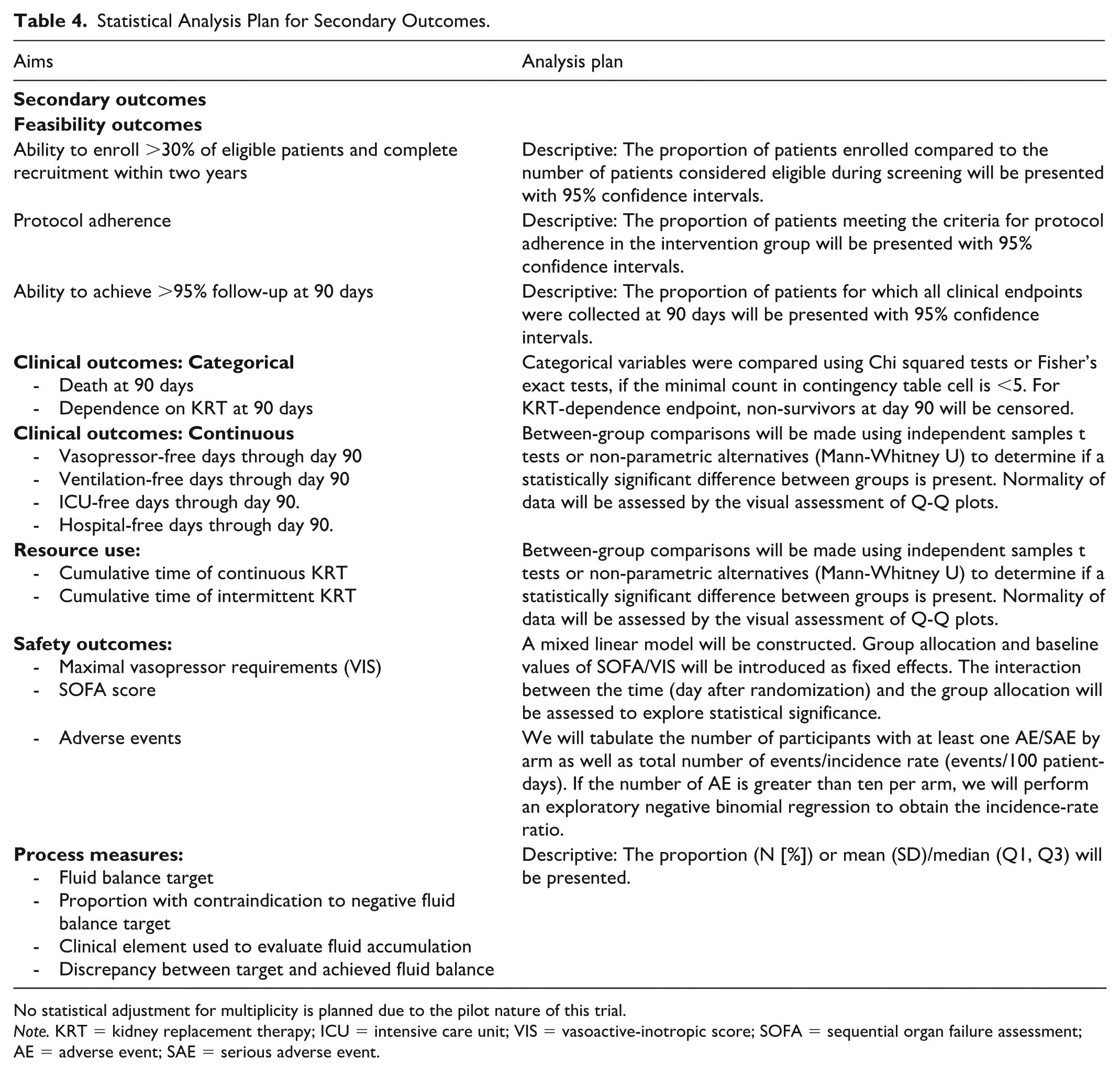
No statistical adjustment for multiplicity is planned due to the pilot nature of this trial.
Note. KRT = kidney replacement therapy; ICU = intensive care unit; VIS = vasoactive-inotropic score; SOFA = sequential organ failure assessment; AE = adverse event; SAE = serious adverse event.
Planned subgroup analyses will be performed according to the initial KRT modality used (intermittent or continuous), sex and gender, cumulative fluid balance from ICU admission to enrolment, urine output in the 24 hours before enrolment, and in patients with sepsis at enrolment.
Trial Oversight
The steering committee includes the leadership team (sponsor principal investigator, co-investigators) and senior investigators with extensive background in critical care nephrology and clinical trials. The committee oversees all operational aspects of the trial, including acquisition and administration of funds, training of research staff, supervision of trial implementation, data interpretation, and completion of the final manuscript. The Applied Health Research Center (AHRC) of Unity Health Toronto serves as the central coordination and data management center.
A data safety and monitoring board (DSMB), consisting of three members with expertise in nephrology, critical care, and trial methodology, meets every six to nine months while the trial is active. The sole mandate of the DSMB is to ensure the safety of the trial’s conduct through the review of all AEs. The DSMB issues reports and makes recommendations regarding trial continuation or modification, which are transmitted to the sponsor, steering committee, and qualified site investigators. This is done according to a predefined DSMB charter (Supplemental Material). No formal interim analyses are planned, but the DSMB has access to data including AEs and data completeness. As of July 2025, two DSMB meetings have already taken place and recommended the continuation of the study.
Ethical Considerations
The trial is being conducted in accordance with the principles of the Good Clinical Practice guidelines. The study was approved by institutional Research Ethics Boards at all participating sites. The trial was registered at clinicaltrials.gov (NCT05473143) prior to the recruitment of the first patient.
Dissemination Policy
The study findings will be presented at national and international meetings and published in peer-reviewed open-access journals. A lay person’s summary of the principal findings of the results will be sent to all patients involved in the study at their request, and we will seek to disseminate this summary as a press release.
Discussion
Defining an optimal fluid-removal strategy after the initiation of KRT is a key research priority to improve the care of critically ill patients with severe AKI. Fluid accumulation has been associated with adverse physiological consequences through congestion and impairment of vital organ function.2-15,39 Despite these potential harms, the clinical implications of fluid management strategies in patients receiving KRT remain largely unknown.
The Probe-Fluid trial is a pilot RCT comparing a protocol-based fluid management strategy with usual care in critically ill patients receiving KRT. The results of this pilot study will inform the feasibility of conducting a definitive multicentre RCT evaluating whether a protocolized fluid management strategy can improve patient-centered outcomes, including mortality and duration of organ support, compared to usual care.
There are limited interventional studies that compare fluid management strategies in critically ill patients undergoing KRT. In the FFAKI multicentre pilot trial,40,41 patients were randomized to initiate protocolized fluid removal at a rate of 1 mL/kg/h, using diuretics or ultrafiltration, less than 12 hours after randomization, or to a control arm in which the initiation of continuous KRT for fluid removal was discouraged. The trial was stopped due to insufficient recruitment after enrolling 23 out of the planned 50 patients. Among those recruited, an important separation between cumulative fluid balance (5814 mL) was noted five days after recruitment. In the more recent single-center trial GONEUTRAL trial, protocolized fluid removal guided by a hemodynamic monitoring protocol was compared to a control group in which minimal net ultrafiltration was allowed, resulting in a significantly lower cumulative fluid balance in the intervention group. 42 In addition, two RCTs, using bioimpedance information to guide fluid removal in critically ill patients receiving KRT,43,44 reported discrepant findings regarding the improvement in bioimpedance-defined achievement of euvolemia, while the impact on cumulative fluid balance was unclear in both trials.
Finally, the RELIEVE-AKI cluster randomized trial compared a restrictive to a liberal ultrafiltration rate in critically ill patients on continuous KRT. 45 For the first six months, all ten participating ICUs were assigned to the liberal ultrafiltration rate group; subsequently, one ICU was randomly allocated to the restrictive group every two months. The liberal group aimed for a net ultrafiltration rate of 2.0 to 5.0 mL/kg/h, while the restrictive group targeted a rate of 0.5 to 1.5 mL/kg/h. A web-based application was used to calculate the hourly ultrafiltration rate range to be set on the KRT machine based on group allocation, the hourly intravenous fluid infusion rate, and the patient’s predicted body weight. The trial was recently stopped after enrolling 99/144 participants. 46
The Probe-Fluid trial was designed with a pragmatic intervention that can be easily scaled to multiple sites without access to special monitoring technology or electronic health records capabilities. It also permits the use of continuous or intermittent KRT modalities according to the center’s usual practices. The proposed intervention was designed to be the least restrictive possible for the participating clinical care teams, accounting for the important heterogeneity in critically ill patients and recognizing that KRT may be initiated in different phases of fluid management, including when a negative fluid balance may not be appropriate. The underlying hypothesis is that the components of the intervention will lead to greater success in preventing and treating fluid accumulation while being associated with minimal risks for the participants. Indeed, since a rigid fluid balance target or net ultrafiltration rate is not mandated as part of the intervention, the clinical care team has the flexibility to tailor the daily fluid management goals to the dynamic clinical course of their patient.
This study has some limitations. Although the intervention is protocolized, its application relies on the clinical judgment of the attending care team, which may lead to variability in adherence across centers. To address this, we have incorporated daily re-evaluation and prescription templates to support consistent implementation. Furthermore, we have included the collection of process measures to acquire a greater understanding of the decisions made by clinicians involved in delivering the intervention. Should the pilot study intervention fail to produce a significant difference in cumulative fluid balance, analysis of process measures will provide the opportunity to consider iterative modifications to our proposed proactive fluid management protocol. Finally, it is not possible to blind the study sites to the nature of the intervention. Awareness of group allocation may influence the attending care team’s decisions regarding co-interventions and aspects of the KRT prescription, introducing potential performance bias. This risk is mitigated by using a standardized, protocolized intervention and objective outcome measures.
Conclusion
The optimal fluid management strategy in critically ill patients with AKI remains unknown. The Probe-Fluid pilot trial is designed to evaluate the feasibility of conducting a definitive multicentre RCT comparing a protocol-based fluid management strategy with usual care in critically ill adult patients receiving KRT.
Supplemental Material
sj-pdf-1-cjk-10.1177_20543581251391877 – Supplemental material for Proactive Prescription-Based Fluid Management Versus Usual Care in Critically Ill Patients on Kidney Replacement Therapy (Probe-Fluid): A Pilot Clinical Trial Protocol
Supplemental material, sj-pdf-1-cjk-10.1177_20543581251391877 for Proactive Prescription-Based Fluid Management Versus Usual Care in Critically Ill Patients on Kidney Replacement Therapy (Probe-Fluid): A Pilot Clinical Trial Protocol by Alicia Shen, Josée Bouchard, Javier A. Neyra, François Lamontagne, Jean-Maxime Côté, Edward G. Clark, Bruno R. da Costa, Martin Gallagher, Neill K. J. Adhikari, Samuel A. Silver, Rita S. Suri, Marlies Ostermann, Ary Serpa Neto, Rinaldo Bellomo, Sean M. Bagshaw, Ron Wald and William Beaubien-Souligny in Canadian Journal of Kidney Health and Disease
Footnotes
Acknowledgements
We thank Dr Kianoush Kashani (Mayo clinic, Rochester, USA), Dr Oleksa Rewa (University of Alberta, Edmonton, Canada), and Dr Etienne Couture (Institut universitaire de cardiologie et de pneumologie de Québec, Québec, Canada) for their contribution to the study as members of the DSMB.
Ethics Approval and Consent to Participate
Informed consent is obtained from all participants or their surrogate decision-maker. The study was approved by institutional research ethics boards (REB) at all participating sites. Ethics approval was obtained from the Center hospitalier de l’Université de Montréal (CHUM) REB (REB 22.118) on 2022-11-10. The study is registered at clinicaltrials.gov (NCT05473143).
Consent for Publication
Not applicable since no patient information is shared.
Availability of Data and Materials
There is no data available for sharing from the work presented in this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The sponsors of the Probe-Fluid trial received funding through a project grant from the Canadian Institutes of Health Research (CIHR-469313). WBS is supported by Fonds de Recherche du Québec: Santé (Clinical Research scholar Junior 1—DOI: 10.69777/312266). JMC is supported by Fonds de Recherche du Québec: Santé (Clinical Research scholar Junior 1—DOI: 10.69777/367766). SMB is supported by a Canada Research Chair in Critical Care Outcomes and Systems Evaluation. JAN is supported by grants from NIH-NIDDK (R01DK128208, R01DK133539, U01DK12998, and U54DK137307).
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: WBS, BRDC, JMC, EGC, JB, MG, NKJA, RW, and AS have no conflict of interest to declare. SAS has received honoraria from Vantive. SMB has received fees for scientific advisory from Baxter, BioPorto, Novartis, Sea Star Medical, and SphingoTec. SMB has received unrestricted grant support from Baxter. JAN has received fees for scientific advisory from Baxter and CovarsaDx. MO received research funding from Baxter and Biomerieux (paid to institution).
Administrative Information
Protocol version: v1.3; February 16, 2023.
Name and contact information for the trial sponsor: William Beaubien-Souligny;
Role of sponsor: Conduct of the study.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.