
Abstract
Background:
Dalteparin sodium, a low-molecular-weight heparin, is indicated for prevention of clotting in the extracorporeal circuit during hemodialysis (HD). Product labeling recommends a fixed single-bolus dose of 5000 international units (IU) for HD sessions lasting up to 4 hours, but adjustable dosing may be beneficial in clinical practice.
Objective:
The aim of the PARROT study was to investigate the safety and efficacy of an adjustable dose of dalteparin in patients with end-stage renal disease requiring 3 to 4 HD sessions per week.
Design:
A 7-week, open-label, multicenter study with a single treatment arm, conducted between October 2013 and March 2016.
Setting:
Ten sites in Canada.
Patients:
A total of 152 patients with end-stage renal disease requiring 3 to 4 HD sessions per week.
Measurements:
The primary outcome was the proportion of HD sessions completed without premature termination due to inadequate anticoagulation.
Methods:
All participants initially received a dose of 5000 IU dalteparin, which could be adjusted at subsequent HD sessions when clinically indicated, by increment or decrement of 500 or 1000 IU, with no specified dose limits.
Results:
Patients were followed for 256 patient-months. Nearly all (99.9%; 95% confidence interval [CI]: 99.7-100) evaluable HD sessions were completed without premature clotting. Dose was adjusted for more than half (52.3%) of participants, mostly owing to clotting or access compression time >10 minutes. Median dalteparin dose was 5000 IU (range: 500-13 000 IU). There were no major bleeds, and minor bleeding was reported in 2.3% of all HD sessions. There was no evidence of bioaccumulation.
Limitations:
This short-term study, with a single treatment arm, was designed to optimize dalteparin dose using a flexible dosing schedule; it was not designed to specifically evaluate dalteparin dose minimization, provide a direct comparison of dalteparin versus unfractionated heparin, or provide information on long-term safety for flexible dalteparin dosing. Patients were excluded if they were at high risk of bleeding, including those on anticoagulants and those on antiplatelet agents other than aspirin <100 mg/d.
Conclusions:
Overall, an adjustable dalteparin sodium dose regimen allowed safe completion of HD, with clinical benefits over fixed dosing.
Trial Registration:
ClinicalTrials.gov NCT01879618, registered June 13, 2013.
What was known before
Anticoagulation is an essential component of hemodialysis (HD) for patients with end-stage renal disease (ESRD), to prevent clotting in the dialyzer and extracorporeal circuit. Dalteparin sodium is a low-molecular-weight heparin (LMWH), which has been approved in Canada since 1994, for the prevention of clotting in the extracorporeal circuit during HD. At the onset of the study, the approved dalteparin dose regimen was 5000 international units (IU), administered intravenously as a single fixed bolus at the start of HD sessions lasting up to 4 hours.
What this adds
The PARROT study investigated the safety and efficacy of an adjustable dose regimen for dalteparin in HD patients with ESRD. The results demonstrated that an adjustable dalteparin dose regimen was well tolerated and efficacious in HD patients with ESRD: Adjustable dosing allowed safe completion of HD. In clinical practice, an adjustable dose regimen may be beneficial to optimally dose and better meet the needs of patients.
Introduction
Anticoagulation is an essential component of hemodialysis (HD) for patients with end-stage renal disease (ESRD), to prevent clotting in the dialyzer and extracorporeal circuit (ECC). Unfractionated heparin (UFH) has traditionally been used to achieve anticoagulation in HD. However, following early adoption in Europe in the 1980s, low-molecular-weight heparins (LMWHs) are increasingly being used worldwide because of clinical reliability, a more predictable anticoagulant effect, longer half-life, ease of administration, and better bioavailability at a low dose.1,2 Although meta-analyses have indicated that LMWHs are as safe and effective as UFH for HD in patients with ESRD,3,4 concerns remain about risks of bleeding and bioaccumulation, 5 despite studies demonstrating no evidence of either.6-10 Consequently, physicians are looking for guidance to optimally dose patients and validate the safety profile of LMWHs for HD. 11
Dalteparin sodium, a LMWH, has been approved in Canada since 1994 for the prevention of clotting in the ECC during HD. 12 The currently approved dose regimen is 5000 international units (IU) administered intravenously as a single fixed bolus at the start of HD sessions lasting up to 4 hours. 12 However, in clinical practice, an adjustable dose regimen may be beneficial to optimally dose and better meet the needs of patients.
The PARROT study was a Phase IIIB Open-Label Study to Optimize the Single Bolus Dose of Dalteparin Sodium for the Prevention of Clotting within the Extracorporeal System During Hemodialysis Procedures for Subjects with Chronic Renal Insufficiency. The aim was to assess the safety and efficacy of an adjustable dalteparin dose regimen to prevent clotting in the ECC during HD sessions in patients with ESRD. All participants initially received a fixed dose of 5000 IU dalteparin, which could be adjusted at subsequent sessions when clinically indicated, by increment or decrement of 500 or 1000 IU. The primary efficacy endpoint was the proportion of HD sessions completed without premature termination due to inadequate anticoagulation.
Methods
Study Design
This was an open-label, multicenter study with a single treatment arm conducted at 10 sites in Canada between October 2013 and March 2016. The trial was conducted in accordance with the Declaration of Helsinki 13 and the International Conference on Harmonization Good Clinical Practice Guidelines. 14 The independent ethics committee at each site approved the study protocol, and all participants provided written informed consent before any procedures were performed (Supplement 1). The study was registered on ClinicalTrials.gov (ID no. NCT01879618). Owing to a typographical error on ClinicalTrials.gov, we documented “clotting in dialyzer” as the primary outcome between June 13, 2013, and November 2, 2016, when it was changed to “mean proportion of successful HD sessions.” The protocol reflected “mean proportion of successful HD sessions” in all versions throughout this period.
Study Population
Eligible participants were adults (18-85 years; >45 kg) with ESRD requiring 3 to 4 HD sessions (⩽4 hours each) per week, with no major intercurrent illness. At screening, eligible participants had received HD for ⩾30 days with only UFH or LMWH (⩽10 000 IU) for anticoagulation and had well-functioning vascular access. Participants of childbearing potential were included provided they were not pregnant or breastfeeding and agreed to use effective contraception through 28 days after last dalteparin dose.
Key exclusion criteria included bleeding disorders (congenital, increased risk, historical, active and uncontrollable, gastrointestinal blood loss, acute gastroduodenal ulcer, hemorrhagic diathesis, cerebral hemorrhage), cancer, thrombophilia, thrombocytopenia, hemoglobin <9.0 g/dL, liver disease, or uncontrolled hypertension (systolic blood pressure ⩾180 mm Hg; diastolic blood pressure ⩾110 mm Hg). Additional exclusions included use of other anticoagulants, antiplatelet therapy (except aspirin <100 mg/d), nonsteroidal anti-inflammatory drugs (except occasional use with proton pump inhibitors), or tissue plasminogen activator; anticipated kidney transplant; hemofiltration; predicted survival <1 year; positive platelet aggregation test with dalteparin; and other conditions for which the use of dalteparin is contraindicated, including diabetic or hemorrhagic retinopathy.
Participants were removed from the study if they experienced major or clinically relevant nonmajor bleeding (Table 1), venous thromboembolism, acute coronary syndrome, or acute cerebrovascular events.
Rating of Bleeding Events.

Note. PRBC = packed red blood cells.
Clinically overt bleeding was defined as bleeding that was directly visible or visible on imaging (eg, ultrasound, computed tomography) or directly observed at surgery, gastroscopy, bronchoscopy, or any other method of visualization.
Interventions
Participants received 3 to 4 HD sessions per week, each lasting up to 4 hours, for up to 20 sessions. All received 5000 IU dalteparin administered as a single bolus into the arterial side of the dialyzer at the beginning of the first HD session. The same dose was continued unless there was an indication for adjustment (Table 2). Dose could be adjusted by 500 or 1000 IU (increment/decrement), dependent on the outcome of the previous HD session and any intervening clinical events, at the discretion of the investigator. Maximal or minimal dose was not restricted.
Clinical Outcomes of the HD Session: Criteria Leading to a Dose Adjustment.

Note. HD = hemodialysis; ECC = extracorporeal circuit; AVF = arteriovenous fistula; AVG = arteriovenous graft.
Assessments and Procedures
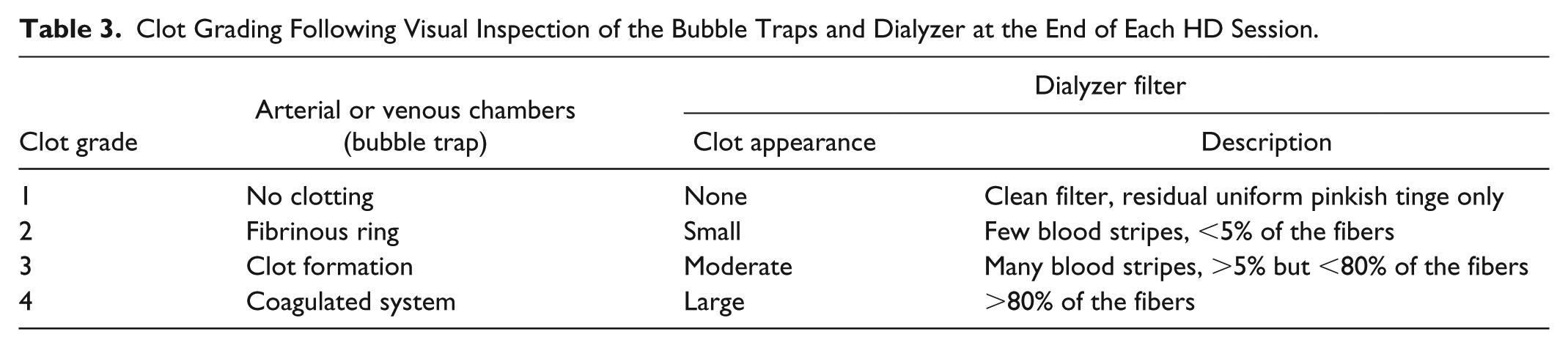
Vital signs (blood pressure, heart rate) and weight (pre- and post-HD) were measured at each visit. Clotting in the ECC, bleeding, access thrombosis, and access compression time (participants with an arteriovenous fistula [AVF] or graft [AVG]) were assessed at each HD session. Clotting in the bubble traps and dialyzer was evaluated by 2 trained individuals by visual inspection, following a 4-point scale (Table 3 and Supplement 2). All research staff received training, based on current standard operating procedures, at the investigator meeting and site initiation visit. Visual inspection was confirmed by a second observer; however, no test-retest reliability was performed. Clinical laboratory tests, including blood biochemistries and hematology, were conducted at screening and end-of-study visits.
Clot Grading Following Visual Inspection of the Bubble Traps and Dialyzer at the End of Each HD Session.
Efficacy
Primary efficacy measures
Any clotting in the bubble traps (arterial and venous chambers) and the dialyzer was evaluated by 2 individuals by visual inspection (Table 3 and Supplement 2). The outcome of each HD session was classified as successful or unsuccessful. A successful session was defined as completion of HD without premature termination due to clotting in the ECC. An unsuccessful session was defined as premature termination (⩾10 minutes before planned) owing to grade 3 or 4 clotting, use of saline flush to prevent clotting, or failure to return the participant’s blood. Note that the occurrence of grade 3 or 4 clotting, use of saline flush, or failure to return blood alone did not result in an HD session being classified as unsuccessful. A session was considered unsuccessful only if any of these occurrences resulted in premature termination. A HD session that terminated prematurely for any other reason was omitted from analyses but was not considered unsuccessful. The primary efficacy endpoint was the mean percent of successful HD sessions.
Secondary efficacy measures
Secondary efficacy endpoints included HD session outcomes and the proportion of HD sessions where the dose was acceptable. Secondary efficacy analyses—comparing fixed versus adjustable dose and dose before versus after adjustment (number of HD sessions with an acceptable dose)—were performed for both primary and secondary endpoints.
Safety
Adverse events (AEs) were assessed at each study visit. Verbatim terms were recorded and coded according to the Medical Dictionary for Regulatory Activities (MedDRA, v19.0). 15 Any AE that was life-threatening or resulted in death, hospitalization, persistent or significant disability/incapacity, or congenital anomaly or birth defect was considered a serious AE (SAE). Lack of efficacy was reported as an AE when associated with an SAE. Access thrombosis was reported if observed during the session.
Bleeding
Bleeding events were assessed at each HD session by the research staff and reported by patients prior to each HD session, as is standard care for all LMWH (Table 1). All reported bleeding events were reviewed at the end of study by the investigators and adjudicated independently. For participants with an AVF or AVG, access compression and access site bleeding times were measured at the end of sessions.
Anti-Xa levels
To determine anti-Xa levels, 3 blood samples were taken (before, and 2 and 4 hours after starting HD [end of HD], at sessions 1, 10, and 20) and analyzed using a validated chromogenic assay at Q2 Solutions (Morrisville, North Carolina). Accumulation of anti-Xa was defined as a trough level of >0.4 IU/mL at the pre-HD measurement before dalteparin administration.
Statistical Analysis of Primary and Secondary Endpoints
For the primary endpoint, the null hypothesis was that the mean proportion of successful HD sessions was ⩽86%. The alternative hypothesis was success rate >86%.
Sample size estimation
The study was considered positive if the 95% confidence interval (CI) lower limit for success rate exceeded 86%. Assuming a success rate of 91%, a sample of 150 participants provided ~84% power to determine whether the study was positive according to simulations of beta-binomial data in which the underlying beta distribution had a mean of 0.91 and an intraclass correlation of 0.30. The sample size incorporated a 5% inflation factor to account for potential attrition of participants from the primary analysis.
Primary analysis
Primary analysis for the primary efficacy endpoint entailed calculation and analysis of the mean percent of successful HD sessions using a generalized estimating equation model for clustered binomial data, with participant as the clustering variable. The study was positive if the 95% CI lower limit was greater than the prespecified value of 86%.
Secondary analysis
Comparative analyses between fixed- and adjustable-dose regimens were performed using a split-sample approach. Two drug administration periods were defined for each participant: period 1 (first HD session through last consecutive session using 5000 IU) and period 2 (first session using an adjusted dose through last session). Fixed dosing was represented by period 1 only, and adjustable dosing by periods 1 and 2.
Participants were randomly split into groups A and B. To provide estimates of success rates, period 1 data were used for group A, and period 1 and 2 data for group B. A permutation resampling approach, which repeatedly and randomly split the sample into groups A and B and calculated the differences in success rates for each compilation, was used to determine a percentile-based 95% CI for the difference in success rates between fixed- and adjustable-dose regimens.
For participants with dose changes, the success rates for periods 1 and 2 were compared using a bootstrap method to estimate a 95% CI for the difference in success rates.
Results
Of 183 HD patients who were screened, 152 were eligible, enrolled, and treated with dalteparin for prevention of clotting in the ECC (Figure 1). Overall, 131 (86.2%) participants completed the study and 21 (13.8%) discontinued prematurely.

Flow of participants through the study.
The study population included 106 (69.7%) men and had a mean age of 57.1 years (Table 4). Median ESRD history was 2.2 years (range: 0.2-25.7), with glomerulonephritis the most common cause (34.9%). All participants had received prior treatment with UFH (88.8%) or LMWH (11.8%), and all received concomitant drug treatments during the study.
Baseline Characteristics for Study Participants.

Note. Data are mean ± SD (range) unless otherwise stated. BMI = body mass index; ESRD = end-stage renal disease; AVF = arteriovenous fistula; AVG = arteriovenous graft; UFH = unfractionated heparin; LMWH = low-molecular-weight heparin; HD = hemodialysis.
Screening visit pre-HD value.
Defined as weight/(height × .01)2 at screening, n = 151.
n = 101.
Participants could have more than one type of access over the course of the study.
Efficacy
Primary efficacy endpoint: Completion of HD without premature termination due to clotting
A total of 2826 HD sessions were performed, of which 2776 (98.2%) were evaluable for the primary efficacy endpoint. Fifty HD sessions were omitted: 20 from one participant who received prohibited medication at HD session 1; 5 for premature termination of HD for reasons other than grade 3 or 4 clotting; 16 for incomplete data; and 9 for not receiving dalteparin. Nearly all (2774 [99.9%; 95% CI: 99.7-100]) evaluable HD sessions were completed without premature interruption due to clotting in the ECC and were considered successful. Two (0.07%) HD sessions were unsuccessful as they terminated early owing to grade 3 or 4 clotting (one session each), both at a dose of 5000 IU. However, grade 3 or 4 clotting did not automatically result in early termination of HD and was recorded in 477 (17.2%) successfully completed sessions. No HD sessions were terminated prematurely because of bleeding.
Secondary efficacy endpoints
HD session outcomes and dose adjustment
Most (114 [75.0%]) participants successfully completed the maximum 20 HD sessions (Figure 1). Depending on the clinical outcome of the HD session (Table 2), dalteparin dose could be adjusted by increment/decrement of 500 or 1000 IU at the next session. Over half (79 [52.3%]) of participants who completed at least one HD session received a dose adjustment, with the remainder (72 [47.7%]) maintaining the fixed 5000 IU dose throughout. Of the 128 (84.8%) participants who completed all 20 HD sessions, more received a dose adjustment (70 [54.7%]) than remained on the standard 5000 IU dose (58 [45.3%]). The proportion of successful sessions was similar for fixed and adjustable dosing periods (–0.1%; 95% CI: –0.3% to 0.1%).
A median of 3 dose adjustments was received by those who completed ⩾1 HD session (range: 1-12) and all 20 sessions (range: 1-10). Dalteparin dose was adjusted for 10% (279) of HD sessions, with grade 3 or 4 clotting at the previous session the most common reason for adjustment (72.8%; Table 5). Dose was generally increased following grade 3 or 4 clotting or use of saline flushes, and decreased following extended access compression time or minor bleeding.
Reasons for Dose Adjustment.

Note. HD = hemodialysis.
Participants could have more than one reason for a dose change.
Three (1.1%) of the “other” dose decreases were due to grade 2 clotting.
Maximal or minimal dose was not restricted, as long as adjustments were made by increments/decrements of 500 or 1000 IU. Median dose was 5000 IU (mean [SD]: 5488 [1191] IU) among all HD sessions (range: 500-13 000 IU; Figure 2). For the 21 patients whose first dose adjustment was a decrease, the subsequent average (SD) dose was 4025 (767) IU, and for the 58 patients whose first dose adjustment was an increase, the average (SD) dose was 6723 (1306) IU. Over half of all HD sessions (1658 [58.9%]) were performed with the standard 5000 IU dose. Dose was increased more frequently than it was decreased, with 888 (31.5%) HD sessions performed above the standard 5000 IU dose compared with 271 (9.6%) below.

Dalteparin sodium doses administered.
Acceptable dose
Each participant completed a mean of 16.1 (SD: 3.79) HD sessions with an acceptable dalteparin dose, ie, no reason for adjustment. Overall, the administered dose was acceptable for 89.8% (2363; 95% CI: 87.4-91.9) of all HD sessions. For the 79 participants whose dose was changed, when moved to an adjustable dose regimen, the proportion of HD sessions with an acceptable dose was 8.3% more than when on a fixed regimen (95% CI: 2.6-16.5).
Safety
Ninety-five (62.5%) participants reported 218 treatment-emergent AEs (TEAEs; Supplement 3). Five (3.3%) participants discontinued the study prematurely because of a TEAE (catheter site bleeding, stoma site bleeding, hematuria, maculopapular rash, and ocular hyperemia, with all apart from catheter site bleeding considered related to dalteparin). Dose changes or temporary discontinuations were reported for 30 (19.7%) participants, most due to coagulation-related TEAEs, which were mostly mild and resolved on the same day. Treatment-emergent SAEs were reported for 3 (2.0%) participants (atrial fibrillation, pneumonia, and influenza) and were not considered treatment related. No deaths were reported.
There were 3 incidents of access thromboses for 2 (of 61; 3.3%) participants with a venous catheter (doses of 5000, 5500, and 6500 IU), and none for participants with an AVF or AVG.
Bleeding events
Of 2818 (99.7%) HD sessions evaluable for safety, 66 (2.3%; 95% CI: 1.6-3.4) were associated with a bleeding event. There were no major bleeds (Tables 1 and 6). One (0.7%; 95% CI: 0.1-3.6) participant reported a clinically relevant nonmajor bleed, and 38 (25.0%; 95% CI: 18.8-32.4) reported one or more minor bleeds. Minor bleeding was reported at 2% (33/1659) of HD sessions performed at 5000 IU and at 10% (10/100) of sessions at 4000 IU.
Bleeding Events and Treatment-Emergent Bleeding-Related AEs.

Note. AEs = adverse events; AVF = arteriovenous fistula; AVG = arteriovenous graft; n = number of participants with AE. Includes data up to 30 days after last dose of study drug; participants are counted only once per treatment in each row.
Sixteen episodes.
Seven episodes.
Thirty-eight episodes.
Non-access-related bleeding—including cuts, hemorrhoid, and conjunctival bleed—was reported for 13 participants. Six (of 61; 9.8%) participants with a central venous catheter reported 7 episodes of bleeding, and 15 (of 91; 16.5%) participants with an AVF reported 38 episodes of access site bleeding, most commonly at the end of a HD session. Access compression time for participants with an AVF was 5 to 10 minutes for over half (57.7%) of all sessions and >10 minutes for 160 (9.0%) sessions. Nine (of 11; 81.8%) participants with an AVG reported 23 episodes of prolonged compression time (>10 minutes).
Anti-Xa levels
Bioaccumulation was assessed by monitoring anti-Xa levels prior to HD at sessions 1, 10, and 20. Predialysis anti-Xa levels fell below the prespecified bioaccumulation threshold (0.40 IU/mL), and below the limit of quantification of the assay (0.04 IU/mL), at all HD sessions for most participants (Figure 3). At HD session 1, the anti-Xa level for one participant (0.55 IU/mL) was above the prespecified bioaccumulation threshold, but below the threshold at subsequent HD sessions. At HD session 10, anti-Xa levels were above the bioaccumulation threshold for 2 (1.3%) participants (0.49 and 0.58 IU/mL). For these 2 participants, anti-Xa levels were below the threshold at HD session 20 (both <0.4 IU/mL), both maintained a fixed dose of 5000 IU dalteparin and reported no bleeding events throughout the study period.

Individual anti-Xa serum levels before HD at sessions 1, 10, and 20.
A total of 1267 samples were assayed for anti-Xa serum levels: 421 of these samples were collected before HD, of which only 9 (2.14%) had anti-Xa levels above the lower limit of quantification for the assay (0.04 IU/mL; Figure 3).
Discussion
Current product labeling for dalteparin sodium as an anticoagulant in the ECC recommends a fixed single dose of 5000 IU for HD sessions lasting up to 4 hours. 12 Limited safety and efficacy data exist for patients who may require lower or higher doses.
The PARROT study investigated the safety and efficacy of an adjustable dalteparin dose regimen. The primary endpoint was met, with nearly all (99.9%) HD sessions completed without premature termination due to clotting. Only 2 HD sessions terminated prematurely owing to grade 3 or 4 clotting, although grade 3 or 4 clotting was reported in 17.2% of sessions that were successfully completed. Of participants who completed at least one HD session, just over half (52%) required a median of 3 dose adjustments, with the majority (76%) involving a dose escalation. The most common reason for dose increase (and adjustment overall) was grade 3 or 4 clotting. Dose was most frequently decreased when access compression time was >10 minutes. Overall, mean (SD) dalteparin dose per HD session was 5488 (1191) IU (range: 500-13 000 IU). The mean (SD) dose for the dose adjustment groups was 4025 (767) IU and 6723 (1306) IU for those whose first adjustment was a decrease or increase, respectively. Dose adjustment resulted in a higher proportion of HD sessions with an acceptable dose compared with fixed dosing.
Similar to our findings, in an open-label, randomized trial evaluating the safety and efficacy of fixed doses of dalteparin or tinzaparin (a LMWH) followed by titration (±500 or 1000 IU), most (91%) of the HD sessions were considered satisfactory or uneventful, with no difference between drug treatment groups. 6 In addition, over half of the dalteparin treatment group required no adjustment from the fixed 5000 IU dose, mean dose was 5546 IU, and the mean number of dose adjustments was 4.9. 6
The PARROT study found that during this 7-week trial of 152 participants receiving either a fixed 5000 IU dose of dalteparin or following dose adjustment (including extreme dose escalation to achieve adequate anticoagulation), major bleeding did not occur, and there was one clinically relevant nonmajor bleed. No HD session was terminated prematurely because of bleeding. Minor bleeding was reported in 2.3% of HD sessions, and access compression time for patients with an AVF was 5 to 10 minutes in over half of the sessions. The trial comparing dalteparin and tinzaparin fixed dose and titration also reported low incidences of major, minor, and extended arteriovenous bleeding (one episode, 1.5% and 3.5%, respectively, for each treatment group). 6 Clinical studies have found no difference in bleeding risk between LMWH and UFH in HD patients.4,16 In addition, a recent study switched 109 HD outpatients previously on UFH to 2500 IU dalteparin, with the option of incremental dose increase (to a maximum of 7000 IU) if clotting was observed. Across all patients, there was no observed increase in bleeding events during 8 months after switching from UFH to dalteparin. 17
A meta-analysis, conducted to evaluate the safety and efficacy of LMWH versus UFH in HD patients, found no difference between anticoagulants (relative risk [RR]: 0.96 [95% CI: 0.27-3.43]). 3 However, limited data were available to evaluate the risk of bleeding, leading to large CIs and therefore limited conclusions. 3 A more recent meta-analysis evaluated the safety of LMWH and UFH for ECC anticoagulation in chronic HD patients. 18 For LMWH, compared with UFH, the RR for total bleeding was 0.76 (95% CI: 0.26-2.22). A total of 55 bleeding events were identified (24 with LMWH, and 31 with UFH), of which 2 were categorized as major bleeding events by the authors (both in the LMWH group, 1 each with enoxaparin and tinzaparin). These data are consistent with no difference in bleeding risk when LMWH is compared with UFH, but larger studies are needed. However, despite these meta-analyses, major bleeding outcomes were not consistently defined or reported in the original studies, and the true number of major bleeding events is unknown.
The use of LMWHs for anticoagulation in the ECC is becoming more prevalent in Canada. Previous trials have demonstrated comparable efficacy and safety of dalteparin and UFH for the prevention of ECC clotting during HD.7,8 In addition, many LMWH products are available in prefilled syringes, which reduce the risk of medication errors, facilitate timely administration, comply with accreditation standards that limit the availability of high-dose UFH formulations in patient care areas, and are cost comparable to vials of UFH.
As there is currently no validated standard threshold to assess anti-Xa bioaccumulation with LMWH, we consulted with thrombosis specialists and nephrologists, and assessed the literature for dalteparin in the clinical setting, during the concept phase of the PARROT study protocol. Previous studies evaluating lower doses of dalteparin (similar to prophylactic LMWH dosing) employed an anti-Xa threshold of 0.4 IU/mL.6,7 In addition, 0.4 IU/mL was used as the threshold in the Dalteparin’s Influence on Renally Compromised: Anti-Ten-A Study (DIRECT) study, which used 5000 IU dalteparin once daily as prophylaxis against venous thromboembolism in critically ill patients with severe renal insufficiency. 19 We therefore selected an anti-Xa threshold level of 0.4 IU/mL pre-dalteparin dose, and at 2 and 4 hours post-dose, to be clinically appropriate for the PARROT study.
Anti-Xa levels were below the predetermined threshold (0.4 IU/mL) before HD for most participants at all HD sessions, indicating no evidence of dalteparin bioaccumulation in the intradialytic period over 20 HD sessions. At HD session 1, ie, prior to receiving dalteparin at the first HD session, the anti-Xa level of 0.55 IU/mL for one participant was surprisingly higher than the threshold for bioaccumulation. The investigators consider this is likely due to a protocol error, where the blood sample for analysis was drawn post-dose, and not pre-dose as instructed. In addition, the elevated anti-Xa levels reported for 2 participants at the beginning of HD session 10 were also likely sampling errors, as levels returned to normal at subsequent sampling (HD session 20), although both participants maintained a dose of 5000 IU throughout the study.
These findings are consistent with other reports using anti-Xa levels as a surrogate marker to measure anticoagulant accumulation of LMWHs.6-10 Two long-term studies have demonstrated no evidence of bioaccumulation with dalteparin when given as an intravenous (IV) bolus followed by continuous IV infusion.7,8 One of these studies, measuring anti-Xa prior to HD, showed no increase in anti-Xa levels during the 6-month dalteparin treatment period (mean values < 0.2 IU/mL). 7 In the second study, anti-Xa levels, measured 2 hours after the start of HD, did not change significantly (values ranged from 0.59-0.74 IU/mL) over the 12-month treatment period. 8 The lack of a control arm in this study prevents direct comparison of dalteparin versus UFH. Although dalteparin dose was decreased for some participants, the study was not designed to specifically evaluate doses below 5000 IU. It is therefore possible that participants who were maintained at 5000 IU could have been successfully treated at a lower dose. Indeed, 2500 IU dalteparin has been effectively used for anticoagulation in the ECC for the majority (>80%) of HD patients in several clinical settings.10,17 In addition, a trial comparing tinzaparin with UFH found that 2500 IU tinzaparin was sufficient for effective anticoagulation in 94% of HD patients. 9 It is possible that a lower dalteparin starting dose may provide a similar efficacy profile and an improved safety profile: Further studies are required to determine effective dose minimization for dalteparin. It is likely that dose increases occurred more frequently than decreases owing to the primary efficacy endpoint, which looked for clotting (resulting in an increase) rather than a stimulus that would result in a decrease, such as bleeding.
A further limitation is the exclusion of some patients at increased risk for bleeding (including those with historical gastrointestinal bleeding, cancer, thrombocytopenia, hemoglobin <9.0 g/dL, liver disease, and on other anticoagulants), who would usually receive anticoagulation for dialysis. Patients with diabetic retinopathy and those on antiplatelet agents other than aspirin <100 mg/d were excluded because of class labeling contraindications: In the clinical setting, they would likely only be excluded from treatment owing to active bleeding. Finally, the present study was a short-term study (20 HD sessions [7 weeks’ treatment]; 256 patient-months from the time of first dose to last follow-up visit), and longer term studies are needed to evaluate the safety of LMWH in chronic HD.
Conclusions
As the use of LMWHs for anticoagulation in the ECC becomes more prevalent in Canada, physicians are seeking guidance and reassurance on how to optimally dose their patients safely while maintaining clinical efficacy. The PARROT study demonstrated that an adjustable dalteparin dose regimen was well tolerated and efficacious in HD patients with ESRD, allowing safe completion of HD.
Supplemental Material
Supplement_1 – Supplemental material for An Adjustable Dalteparin Sodium Dose Regimen for the Prevention of Clotting in the Extracorporeal Circuit in Hemodialysis: A Clinical Trial of Safety and Efficacy (the PARROT Study)
Supplemental material, Supplement_1 for An Adjustable Dalteparin Sodium Dose Regimen for the Prevention of Clotting in the Extracorporeal Circuit in Hemodialysis: A Clinical Trial of Safety and Efficacy (the PARROT Study) by Steven Soroka, Mohsen Agharazii, Sandra Donnelly, Louise Roy, Norman Muirhead, Serge Cournoyer, Martin MacKinnon, Neesh Pannu, Brendan Barrett, François Madore, Karthik Tennankore, Jo-Anne Wilson, Fiona Hilton, Nancy Sherman, Kevin Wolter, John Orazem and Guillaume Feugère in Canadian Journal of Kidney Health and Disease
Supplemental Material
Supplement_2 – Supplemental material for An Adjustable Dalteparin Sodium Dose Regimen for the Prevention of Clotting in the Extracorporeal Circuit in Hemodialysis: A Clinical Trial of Safety and Efficacy (the PARROT Study)
Supplemental material, Supplement_2 for An Adjustable Dalteparin Sodium Dose Regimen for the Prevention of Clotting in the Extracorporeal Circuit in Hemodialysis: A Clinical Trial of Safety and Efficacy (the PARROT Study) by Steven Soroka, Mohsen Agharazii, Sandra Donnelly, Louise Roy, Norman Muirhead, Serge Cournoyer, Martin MacKinnon, Neesh Pannu, Brendan Barrett, François Madore, Karthik Tennankore, Jo-Anne Wilson, Fiona Hilton, Nancy Sherman, Kevin Wolter, John Orazem and Guillaume Feugère in Canadian Journal of Kidney Health and Disease
Supplemental Material
Supplement_3 – Supplemental material for An Adjustable Dalteparin Sodium Dose Regimen for the Prevention of Clotting in the Extracorporeal Circuit in Hemodialysis: A Clinical Trial of Safety and Efficacy (the PARROT Study)
Supplemental material, Supplement_3 for An Adjustable Dalteparin Sodium Dose Regimen for the Prevention of Clotting in the Extracorporeal Circuit in Hemodialysis: A Clinical Trial of Safety and Efficacy (the PARROT Study) by Steven Soroka, Mohsen Agharazii, Sandra Donnelly, Louise Roy, Norman Muirhead, Serge Cournoyer, Martin MacKinnon, Neesh Pannu, Brendan Barrett, François Madore, Karthik Tennankore, Jo-Anne Wilson, Fiona Hilton, Nancy Sherman, Kevin Wolter, John Orazem and Guillaume Feugère in Canadian Journal of Kidney Health and Disease
Footnotes
Authors’ Note
Coauthor Irina Kaplan died in January 2017. The data in this article have been presented in poster format at the Canadian Society of Nephrology (CSN 2017), May 4-6, 2017, Montreal, Canada, and European Renal Association – European Dialysis and Transplant Association (ERA-EDTA 2017), June 3-6, 2017, Madrid, Spain. Editorial support was provided by Alexandra Bound, PhD, of Engage Scientific Solutions, London, UK, and was funded by Pfizer.
Availability of Data and Materials
Upon request, and subject to certain criteria, conditions and exceptions (see ![]() for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Ethics Approval and Consent to Participate
The independent ethics committee at each site approved the study protocol, and all participants provided written informed consent before any procedures were performed (Supplement 1).
Consent for Publication
Consent for publication was obtained from all authors.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: SS, MA, SD, LR, NM, SC, MM, NP, BB, FM, and KT have no relationship with Pfizer other than being a site investigator in the PARROT study. JW has received consultancy fees from Pfizer in the past 3 years. FH, NS, KW, JO, and GF are full-time employees of Pfizer Inc and hold stock and/or stock options.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Pfizer. The study sponsor was involved in the design of this study and its execution, analyses, interpretation of the data, and decision to submit results.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.