
Abstract
Purpose:
The purpose of this article is to update the previously published consensus recommendations from March 2017 discussing the optimal management of adult patients with autosomal dominant polycystic kidney disease (ADPKD). This document focuses on recent developments in genetic testing, renal imaging, assessment of risk regarding disease progression, and pharmacological treatment options for ADPKD.
Sources of information:
Published literature was searched in PubMed, the Cochrane Library, and Google Scholar to identify the latest evidence related to the treatment and management of ADPKD.
Methods:
All pertinent articles were reviewed by the authors to determine if a new recommendation was required, or if the previous recommendation needed updating. The consensus recommendations were developed by the authors based on discussion and review of the evidence.
Key findings:
The genetics of ADPKD are becoming more complex with the identification of new and rarer genetic variants such as GANAB. Magnetic resonance imaging (MRI) and computed tomography (CT) continue to be the main imaging modalities used to evaluate ADPKD. Total kidney volume (TKV) continues to be the most validated and most used measure to assess disease progression. Since the publication of the previous consensus recommendations, the use of the Mayo Clinic Classification for prognostication purposes has been validated in patients with class 1 ADPKD. Recent evidence supports the benefits of a low-osmolar diet and dietary sodium restriction in patients with ADPKD. Evidence from the Replicating Evidence of Preserved Renal Function: an Investigation of Tolvaptan Safety and Efficacy in ADPKD (REPRISE) trial supports the use of ADH (antidiuretic hormone) receptor antagonism in patients with ADPKD 18 to 55 years of age with eGFR (estimated glomerular filtration rate) of 25 to 65 mL/min/1.73 m2 or 56 to 65 years of age with eGFR of 25 to 44 mL/min/1.73 m2 with historical evidence of a decline in eGFR >2.0 mL/min/1.73 m2/year.
Limitations:
Available literature was limited to English language publications and to publications indexed in PubMed, the Cochrane Library, and Google Scholar.
Implications:
Advances in the assessment of the risk of disease progression include the validation of the Mayo Clinic Classification for patients with class 1 ADPKD. Advances in the pharmacological management of ADPKD include the expansion of the use of ADH receptor antagonism in patients 18 to 55 years of age with eGFR of 25 to 65 mL/min/1.73 m2 or 56 to 65 years of age with eGFR of 25 to 44 mL/min/1.73 m2 with historical evidence of a decline in eGFR >2.0 mL/min/1.73 m2/year, as per the results of the REPRISE study.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is characterized by bilateral renal cysts that destroy normal tissue as they grow, leading to renal fibrosis, renal architectural derangement, and ultimately renal failure.1,2 There are currently no validated estimates of the prevalence of ADPKD in Canada. In a recent meta-analysis of European literature, the prevalence of ADPKD in Europe was reported to be 2.7 per 10 000. 3 This prevalence is likely similar to that in the Canadian population despite no formal data. Recent population-based minimum point ADPKD estimates of 2.9 and 3.3 per 10 000 have been calculated, accounting for 7% to 11% of patients on renal replacement therapy in Europe and approximately 5% of patients requiring dialysis in the United States.2,4,5 Approximately 45% to 70% of patients with ADPKD progress to end-stage renal disease (ESRD) by 65 years of age. 6 Targeted pharmacological therapies that have been tested in the treatment of ADPKD include mammalian target of rapamycin (mTOR) inhibitors, somatostatin analogues, tyrosine kinase inhibitors, and a vasopressin V2 receptor antagonist.4,7
The goal of this update is to provide nephrologists and other health care practitioners with updated recommendations on assessing the risk of disease progression and pharmacological management of patients with ADPKD based on evidence published since the development of the first consensus recommendation. In April 2016, a panel of nephrologists from across Canada met to develop evidence-informed recommendations for the optimal management of adult patients with ADPKD, with a focus on the role of genetic testing, renal imaging, risk-prediction of disease progression, and pharmacological treatment options. The recommendations were published in the March 2017 issue of the Canadian Journal of Kidney Health and Disease. 8 The updated recommendations published herein serve to update these same topics.
Methods
The present updated consensus recommendations are based on the experience and opinions of the authors, and on a literature search conducted in PubMed, the Cochrane Library, and Google Scholar using the search terms “ADPKD” or “polycystic kidney” in combination with the following terms: “CKD” or “chronic kidney disease” or “diagnosis” or “end-stage renal disease” or “ESRD” or “gene” or “imaging” or “management” or “mTOR inhibitor” or “risk” or “pharmacological” or “screening” or “somatostatin” or “surgery” or “TKV” or “total kidney volume” or “height adjusted TKV” or “tolvaptan” or “transplantation” or “treatment.” We selected publications that were published from January 2016 to January 2018 to identify the latest evidence published since the April 2016 meeting. All identified papers were reviewed by the authors and consensus was reached on which papers did not have sufficient information relevant or within scope for this update. The papers had to provide validation for previous recommendations or new information that would change a previous recommendation or result in a new recommendation. A total of 43 publications were identified, of which 21 contained information relevant to this update. The authors reached a consensus on the updated recommendations published herein. Consensus was achieved by discussion of the evidence and focusing on the scope of the recommendations. No disputes had to be settled by vote or other means. The following aspects of the disease, addressed in the previous recommendations, were updated: genetic testing, renal imaging, predicting disease progression, and pharmacological treatment options.
Identifying Patients With ADPKD
Since the previous consensus recommendations for the optimal management of adult patients with ADPKD were drawn up, other groups have published guidelines and a consensus report designed to address clinical challenges and offer guidance in the management of patients with ADPKD.8,9 Offering treatment early to these patients slows cyst growth, thereby delaying both loss of kidney function and the need for renal replacement therapy, as well as improving patients’ quality of life.
We suggest that all patients with a diagnosis of ADPKD or suspected ADPKD be referred to a nephrologist for initial assessment. Initial assessment should include kidney imaging and, in some cases, genetic testing to determine the patient’s risk of rapid progression and to determine what treatment should be initiated.
Genetic Testing in ADPKD
The majority of mutations in ADPKD occur in the PKD1 and PKD2 genes which encode two proteins, polycystin-1 and polycystin-2, that constitute the transient receptor potential polycystin subfamily of transient receptor potential channels.10-12 Rarer variants such as GANAB, which encodes the glucosidase IIα subunit, have recently been discovered and research into these novel mutations is ongoing.13-15 Approximately 10% of patients have no identifiable mutation; these patients usually have a minor ADPKD phenotype. 13 With more than 2000 mutations having been identified to date, the genetics of ADPKD are becoming more and more complex (Autosomal Dominant Polycystic Kidney Disease Mutation Database [PKDB], http://pkdb.mayo.edu/).
Genetic testing for ADPKD can be carried out using deoxyribonucleic acid (DNA) linkage analysis, 16 gene-based mutation screening (also referred to as Sanger sequencing), 17 or next-generation sequencing (NGS). 18 Genetic testing is not indicated for all patients with ADPKD. For example, it is not needed when a firm positive or negative diagnosis can be made by imaging alone, when a diagnosis can be made based on the imaging results of the patient’s parents, or when a diagnosis can be made based on the presence of extrarenal manifestations. Genetic testing, however, may be considered to confirm the absence of any mutations for ADPKD in living relatives who are potential donors, to identify mutations in patients without a family history of ADPKD, to exclude other cystic kidney diseases in families with atypical radiographic patterns of kidney cysts, to identify mutations in families affected by early-onset polycystic disease, and to offer prenatal or preimplantation diagnosis.16,19 There is wide variability in access to genetic testing in Canada at this time. If genetic testing is to be done, it should be coordinated through a center with the appropriate expertise and interpreted by those familiar with ADPKD genetics.
Based on existing data, genetic testing is not necessary for selecting treatment options for patients with a confirmed diagnosis of ADPKD. If genetic testing is to be done, it should be performed by a Clinical Laboratory Improvement Amendments (CLIA)–certified center and interpreted by those familiar with ADPKD genetics.
Based on existing data, genetic testing is not necessary for selecting treatment options for patients with a confirmed diagnosis of ADPKD.
Renal Imaging
Imaging modalities used to diagnose and evaluate ADPKD include abdominal ultrasound (US), computed tomography (CT), and magnetic resonance imaging (MRI). MRI is the preferred modality for kidney size determination due to its greater accuracy and precision compared with US. 20 CT performs well; however, it exposes patients to radiation. 21 Compared with MRI or CT, US is more practical and more cost-effective. 22 However, it is difficult to obtain accurate and reproducible results with US, as it is more dependent on the proficiency of the technician than MRI or CT.
In ADPKD, total kidney volume (TKV) continues to be the most validated and most used measure to assess disease progression. Not only are changes in overall TKV less variable than changes in kidney cystic and noncystic components, height-adjusted TKV (htTKV) is easier to obtain than other measures.23-26 TKV may be determined by stereology or other formulae, such as the ellipsoid equation, that estimate volume using a limited set of measurements.23,24 TKV may also be determined using automated methods that classify patients based on htTKV and age.25,26
1. We recommend that before quantifying the size of the kidneys, patients should be classified according to the Mayo Clinic classification for typical versus atypical morphology with renal imaging. 2.1 We recommend that a baseline assessment of renal size be undertaken in patients with ADPKD. The objective of these measurements is to determine which patients are suitable candidates to be considered for therapeutic intervention based on their risk of progression. 2.2 Although the gold standard for measuring TKV is MRI stereology, we recommend the use of ellipsoid TKV or US to determine TKV in routine clinical practice. We suggest that MRI or CT htTKV is currently the most accurate method of assessing renal size in patients with ADPKD. 2.3 In the absence of MRI, imaging by CT may be used to determine TKV. In situations where an MRI or CT is not easily obtainable, we suggest US-measured kidney length (KL) as a suitable surrogate. US can be used to determine TKV; however, TKV obtained using US may introduce error and does not provide an advantage over KL. 3. We recommend that routine assessment of TKV or KL should not exceed a frequency of once yearly.
Imaging in ADPKD can be classified into 2 categories: renal imaging for diagnosis, and renal imaging for prognosis and disease progression. The preferred method for confirming the presence of ADPKD in patients with a family history is US imaging and the use of the Unified Criteria (Table 1). 27 In the absence of family history, there are no validated criteria to diagnose ADPKD based on US imaging. In select circumstances (eg, lack of family history, younger age), the diagnosis of ADPKD may require higher sensitivity imaging, including assessing extrarenal manifestations, and genetic testing. Renal imaging can be used to characterize ADPKD as typical or atypical presentation using the Mayo Clinic classification (Table 2 and Figure 1). 24 According to this system, patients with typical symmetric, bilateral, diffuse cyst distribution are categorized as class 1 (approximately 90% of patients), and patients with atypical, asymmetric, or segmental cyst distribution are categorized as class 2. In addition to the Mayo Classification, well-established Unified Criteria for using US to diagnose ADPKD have been in use for several years (Table 1).27,28
Unified Criteria for Ultrasound Diagnosis of ADPKD. 27

Note. All values presented are mean estimates. ADPKD = autosomal dominant polycystic kidney disease; PPV = positive predictive value; SEN = sensitivity.
Unilateral or bilateral.
Classification of ADPKD Based on Imaging Characteristics According to the Mayo Clinic Classification. 24

Source. Republished from Irazabal et al 24 with permission of the American Society of Nephrology; permission conveyed through Copyright Clearance Center, Inc.
Note. ADPKD = autosomal dominant polycystic kidney disease; TKV = total kidney volume.
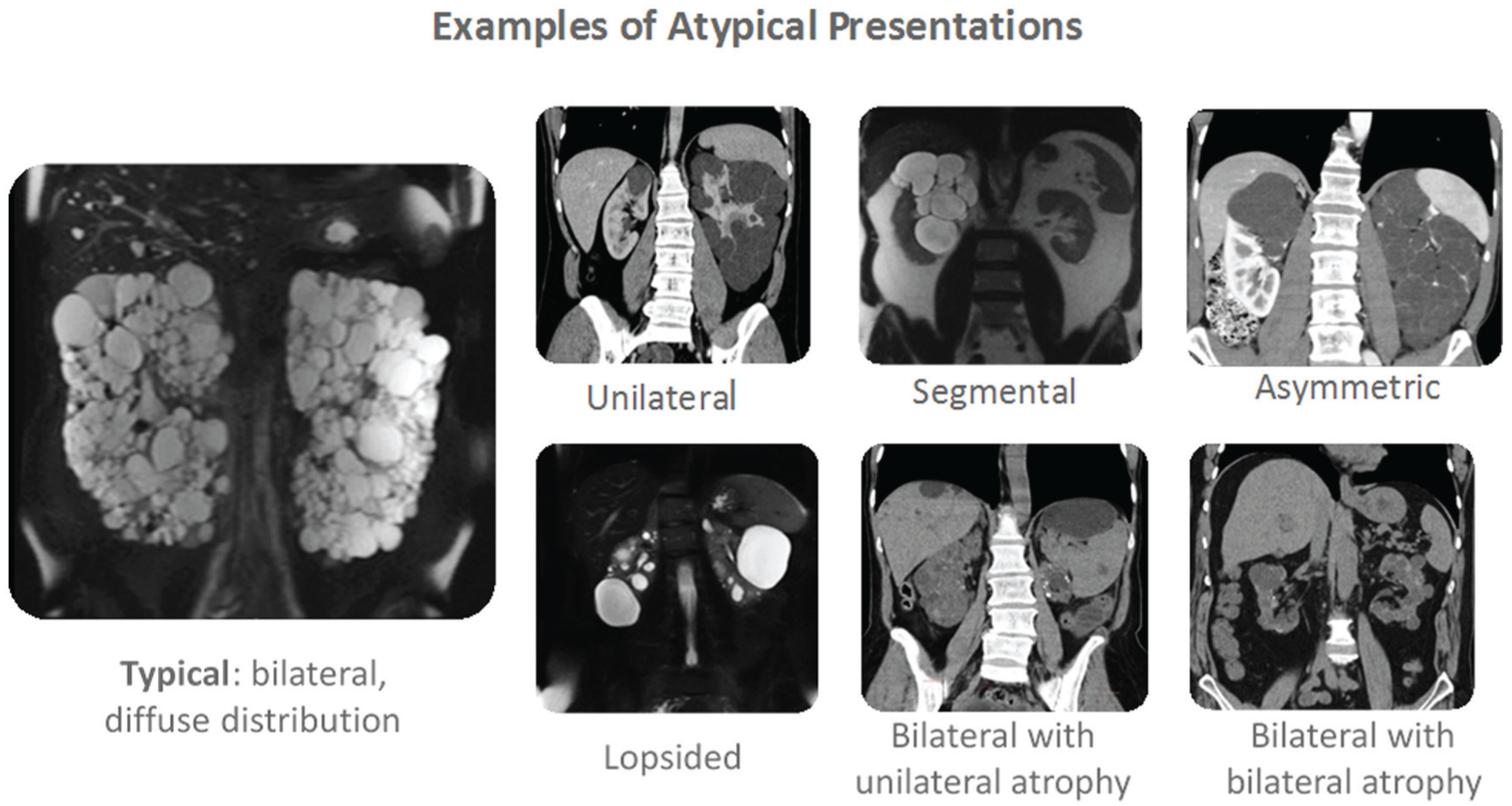
Mayo Clinic classification of autosomal dominant polycystic kidney disease. 24
The preferred method for confirming the presence of ADPKD in patients with a family history is US imaging and the use of the Unified Criteria to establish diagnosis and determine if typical or atypical.
In select circumstances, such as in patients without a family history of ADPKD, other imaging modalities, including CT or MRI, may be considered for diagnosing ADPKD, particularly to detect cysts in younger patients.
When imaging for prognosis in ADPKD, several considerations must be taken into account. Although MRI stereology is the gold standard for measuring TKV, we suggest that ellipsoid TKV or US be used to determine TKV in routine clinical practice. We believe that MRI or CT htTKV are currently the most accurate methods for the assessment of renal size in patients with ADPKD. When MRI imaging is not available, we believe CT imaging may be used to measure TKV. If both MRI and CT imaging are not available, we believe using US-measured KL to be a suitable surrogate. US can be used to determine TKV; however, TKV obtained using US may introduce error and does not provide an advantage over KL. We also believe general abdominal imaging using MRI is useful in obtaining baseline images of the liver, spleen, and pancreas to monitor changes in hepatic cysts, pancreatic enlargement and/or cysts, and splenic enlargement and/or cysts. Finally, in younger patients (<40 years of age) with a normal US, MRI imaging may be required to confirm the presence of disease.
Renal imaging, along with the Mayo Clinic Classification, may also be used for prognostication purposes. 24 By integrating htTKV with age, this system divides class 1 patients into subclasses A through E, where classes 1C, 1D, and 1E show the highest propensity for developing early-onset renal disease (Figure 2a). Class 2 patients are generally not at risk of rapid disease progression. Once patients are classified according to the Mayo Clinic criteria, their disease progression over time may be predicted (Figure 2b). The Mayo Clinic Classification tool is available at http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754.

(a) Subclassification of patients with class 1 autosomal dominant polycystic kidney disease at baseline based on htTKV and age at baseline; (b) predicted change in eGFR over time in class 1 patients (slopes shown are those for men). 24
When the first consensus recommendations were published, the Mayo Clinic Classification had not been validated in the clinical setting. Although it was believed to be a useful clinical tool, it had been developed as a way to identify patients eligible to participate in clinical trials. Therefore, its utility in a broader patient population was unknown. Since then, Girardat-Rotar et al validated the use of the Mayo Clinic Classification in patients with class 1 ADPKD in the clinical setting using data collected from 2006 to 2016 from the ongoing Swiss ADPKD study. 29
Finally, renal imaging may be used to monitor a patient’s disease progression. There continue to be limited data on the role of repeated imaging in the management of patients with ADPKD.20,30 Studies have shown that serial measurements can detect an increase of >5% per year in TKV, which corresponds to the threshold for class 1D and appears to correlate well with predicting rapid disease progression. 24 Recommendations published by the European Renal Association – European Dialysis and Transplant Association (ERA-EDTA) and the Japanese regulatory authorities state that patients showing an annual increase of >5% in TKV should be categorized at higher risk for disease progression.8,31 Recently, results published from the extension of the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease study (CRISP III) support the use of serial TKV measurements to monitor disease progression in ADPKD. 32 This study showed that the rate of change in htTKV was negatively correlated with change in glomerular filtration rate (GFR), thereby demonstrating the prognostic value of serial TKV measurements over time.
We recommend that a baseline assessment of renal size be undertaken in patients with ADPKD. The objective of these measurements is to determine which patients are at risk of rapid progression and may be suitable candidates to be considered for therapeutic intervention based on their risk of progression.
In patients with typical morphology, we recommend using US to measure KL, or MRI or CT to measure TKV (and to calculate htTKV where appropriate) if a more precise measurement is required for therapeutic decisions. In cases where historical images are available, those images should be consulted before requesting new imaging.
After a baseline assessment of renal size, not all patients require routine reassessment of renal size. If renal size reassessment is performed, we recommend it should not exceed a frequency of once yearly.
Assessing Risk of Rapid Disease Progression
In ADPKD, disease progression can be monitored by measuring TKV, creatinine clearance, or estimated glomerular filtration rate (eGFR) using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation.24,33 In the early stages of ADPKD, hyperfiltration serves to maintain renal function such that changes in renal function are difficult to observe. In contrast, changes in TKV are readily observable, making it a more sensitive marker in this stage of the disease process. 23 Prognostic factors for disease progression in ADPKD are summarized in Table 3. Risk for disease progression can be predicted using predicting renal outcomes in ADPKD (PROPKD) scoring, genetic scoring, or Mayo Clinic Classification.24,34
Prognostic Factors Related to Disease Progression in ADPKD.

Note. ADPKD = autosomal dominant polycystic kidney disease; TKV = total kidney volume; GFR = glomerular filtration rate; HtTKV = height-adjusted total kidney volume; ESRD = end-stage renal disease; PPV = positive predictive value; eGFR = estimated glomerular filtration rate; TEMPO = Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes; NGAL = neutrophil gelatinase-associated lipocalin; IL = interleukin; CRISP = Consortium of Renal Imaging Studies in Polycystic Kidney Disease; BP = blood pressure.
Although testing for NGAL and IL-18 is carried out in the research setting, these tests are not readily available, and therefore not measured, in routine clinical practice.
Risk Prediction Using PROPKD Score
The PROPKD scoring system predicts renal outcomes in patients with ADPKD on the basis of genetic and clinical data from 1341 patients from the Genkyst study cohort. 34 This scoring system assigns points as shown in Table 4, with total scores ranging from 0 to 9 points. The scoring system assigns patients to 1 of 3 risk categories, with corresponding predicted median age of onset for ESRD and predicted disease progression (Table 5). It should be noted that the PROPKD scoring system cannot be applied to patients with no history of urological events or hypertension. Since the publication of the first consensus recommendations, the prognostic value of the PROPKD scoring system has been confirmed in a post hoc analysis of the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes (TEMPO) 3:4 trial. 45
The predicting renal outcomes in ADPKD (PROPKD) Scoring System. 34

Source. Republished from Cornec-le Gall et al 34 with permission of the American Society of Nephrology; permission conveyed through Copyright Clearance Center, Inc.
Predicted Median Age of Onset of ESRD and Predicted Disease Progression by predicting renal outcomes in ADPKD (PROPKD) Risk Category. 34 .
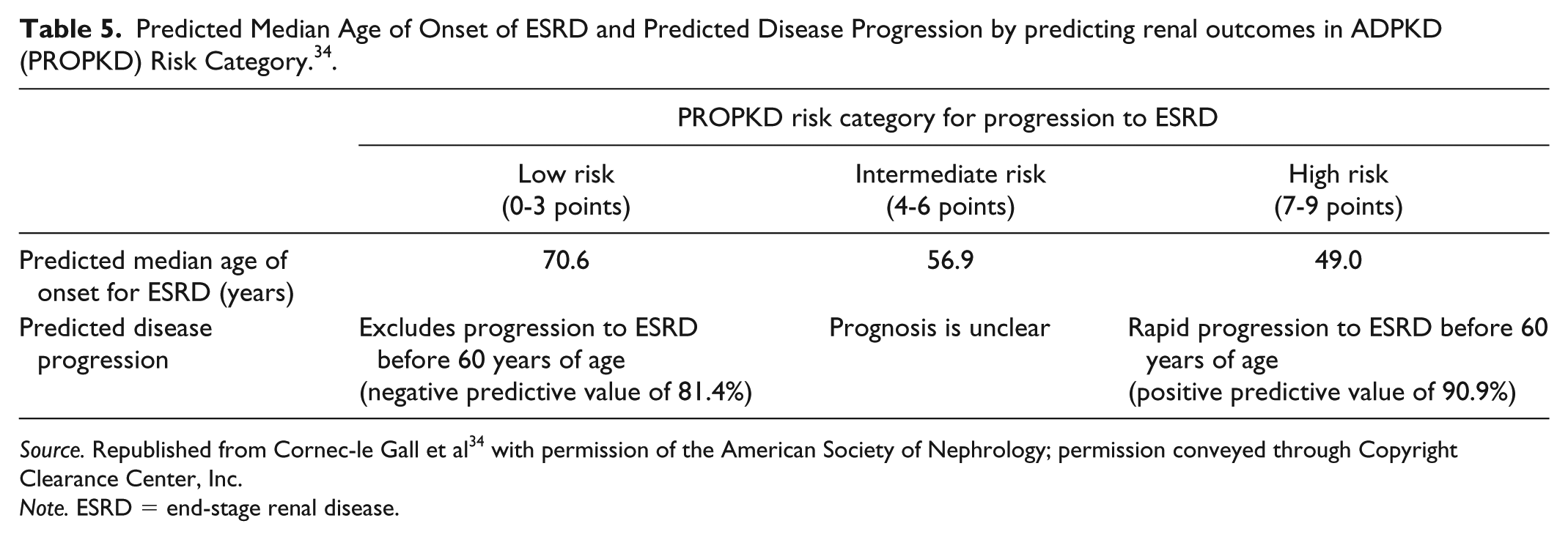
Source. Republished from Cornec-le Gall et al 34 with permission of the American Society of Nephrology; permission conveyed through Copyright Clearance Center, Inc.
Note. ESRD = end-stage renal disease.
Risk Prediction Using Genetic Scoring
The PROPKD scoring system has lower accuracy in some patients, such as those younger than 35 years and those missing clinical data. 34 In these cases, genetic scoring may be carried out based on gender and genetic data. With this scoring system, patients fall into 1 of 4 prognostic groups:
patients with PKD2 mutations (1 point)
patients with nontruncating PKD1 mutations (2 points)
women with truncating PKD1 mutations (3 points)
men with truncating PKD1 mutations (4 points)
Patients with a genetic score ⩾2 points have a predicted onset of ESRD before age 65 years. Although genetic scoring is less accurate than the PROPKD score, it offers good prediction of ESRD.
Risk Prediction Using Mayo Clinic Classification
As discussed in the previous section, the Mayo Clinic Classification defines groups of patients with different risks for eGFR decline based on htTKV and age at baseline (Figure 2a), which, in turn, predicts decline in eGFR (Figure 2b). 24 Although this classification system has been externally validated, 29 its application to clinical practice has not yet been delineated.
We recommend that in current clinical practice, patients with a TKV measurement be categorized in terms of their risk of progression as per the Mayo Clinic Classification or other validated clinical tools. We highlight that the application of the Mayo Clinic Classification to clinical practice has not yet been delineated; however, it appears to be the most robust clinical prediction tool as it pertains to the important marker of htTKV. Currently available TKV-based prognostication tools should not be applied to class 2 (atypical morphology) patients, as we suggest that these patients are unlikely to be rapid progressors. Certain patients may require further clinical evaluation. We suggest that patients who are classified as Mayo class 1C, D, or E be considered to be at risk of rapid progression of their ADPKD renal disease. We recommend that patients who demonstrate a sequential increase of >5% annually in TKV on imaging should be considered at risk of rapid progression of their ADPKD-related renal disease. We recommend that patients with an US KL of >16.5 cm bilaterally should be considered at high risk of progression of their ADPKD-related renal disease. A KL >16.5 cm has been shown to correlate with a TKV of 750 mL; however, direct measurement of TKV would be required if more accurate assessment is needed. We suggest that baseline TKV or KL are important determinants of renal progression of ADPKD; however, serial TKV or KL measurements have not been established as markers to monitor response to therapy.
We recommend that in current clinical practice, patients with an htTKV measurement be categorized in terms of their risk of progression as per the Mayo Clinic Classification or other validated clinical tools. Currently available TKV-based prognostication tools should be applied only to class 1 (typical morphology) patients, as we suggest these patients are likely to be rapid progressors. Certain patients may require further clinical evaluation. We suggest that patients be considered at risk of rapid progression of ADPKD renal disease if they meet either of the following criteria: (1) classified as Mayo class 1C, D, or E, or (2) have an US KL of >16.5 cm bilaterally. We suggest the following be used as markers of rapid progression: (1) a sequential increase of >5% annually in htTKV on imaging, or (2) documented disease progression (eg, rapid decline in eGFR; defined as decline in eGFR >2.5 mL/min/1.73 m2 annually; patients in the placebo group of the TEMPO 3:4 study showed an annual decline in eGFR of 3.5 mL/min/1.73 m2 over the three years of observation).
Nontargeted Treatment Options
Nontargeted treatment options for ADPKD discussed in the previous consensus recommendations included protein restriction, increased fluid intake, and blood pressure (BP) control. To date, no compelling evidence supports the use of protein restriction and increased fluid intake as nontargeted treatment options.34,46 Rigorous BP control, however, significantly reduces urinary albumin excretion and produces a significantly lower annual rate of increase in TKV.34,47
Recently, several studies have evaluated the effects of low-osmolar diet, coffee consumption, and dietary sodium restriction on disease progression in ADPKD.48-50 Combining a low-osmolar diet with adjusted water intake to achieve a urine osmolality of ⩽280 mOsm/kg water has shown to produce significantly reduced vasopressin secretion in patients with ADPKD compared with no intervention. 48 After 2 weeks, mean (standard deviation [SD]) plasma copeptin levels decreased significantly from 6.2 ± 3.05 pmol/L to 5.3 ± 2.5 pmol/L (P = .02) in patients that combined a low-osmolar diet with adjusted water intake, while levels did not change (from 4.7 ± 3.6 to 5.07 ± 4 pmol/L; P = .2) in a control group of patients with no intervention. The change in copeptin levels between the 2 groups was significant (−0.86 ± 1.3 pmol/L vs +0.39 ± 1.2 pmol/L, respectively, P = .009). Mean (SD) urine osmolality decreased from 426 ± 193 mOsm/kg water to 258 ± 117 mOsm/kg water (P = .01) in the intervention group, and did not change (from 329 ± 159 mOsm/kg water to 349 ± 139 mOsm/kg water; P = .3) in the control group. The change in urine osmolality was significant between the 2 groups (−167 ± 264 mOsm/kg water vs +20 ± 80 mOsm/kg water, respectively, P = .007). The dietary intervention led to a significant reduction in vasopressin secretion and a reduction in the amount of water required to reduce vasopressin secretion.
A prospective study designed to assess the association between coffee consumption and disease progression in patients with ADPKD showed that coffee drinkers did not have a statistically significant difference in kidney size (difference of −33.03 cm3 htTKV, P = .10), and did not have a statistically significant difference in eGFR (2.03 mL/min/1.73 m2, P = .089), compared with noncoffee drinkers after multivariate adjustment for age, smoking, hypertension, sex, body mass index, and an interaction term (Coffee × Visit). 49 This study is the first to demonstrate that coffee drinking does not adversely affect kidney size or function in patients with ADPKD.
A post hoc analysis of the Polycystic Kidney Disease Treatment Network (HALT-PKD) trials (study A and study B) showed that dietary sodium restriction was beneficial in the management of patients with ADPKD. 50 Briefly, study A randomized hypertensive patients with ADPKD (15-49 years of age, with eGFR >60 mL/min/1.73 m2) to either a standard BP target of 120/70 to 130/80 mm Hg or a low BP target of 95/60 to 110/75 mm Hg, and to either treatment with the angiotensin-converting enzyme inhibitor lisinopril plus the angiotensin-receptor blocker telmisartan or treatment with lisinopril plus placebo. 51 Study B randomized patients with ADPKD (18-64 years of age, with eGFR 25-60 mL/min/1.73 m2) to either treatment with lisinopril plus telmisartan or lisinopril plus placebo, with dose adjustments to achieve a BP between 110/70 and 130/80 mm Hg. 52 All patients were instructed to follow a sodium-restricted diet of ⩽2.4 g/day (⩽100 mmol/day). 50 In study A, mean urinary sodium excretion decreased significantly by 0.25 mmol/24 h per month and by 0.41 mmol/24 h per month in study B. In study A, averaged urinary sodium excretion and time varying urinary sodium excretion were significantly associated with kidney growth at 0.43%/year and 0.09%/year, respectively, for each 18 mmol urinary sodium excretion. In study B, averaged urinary sodium excretion was significantly associated with increased risk for the composite endpoint of time to death, ESRD, or a 50% reduction from the baseline eGFR (hazard ratio = 1.08 for each 18 mmol/24 h increase in urinary sodium excretion, P = .010) and significantly associated with faster annual eGFR decline (−0.086 mL/min/year for each 18 mmol/24 h increase in urinary sodium excretion). Current guidelines published by Hypertension Canada suggest reducing sodium intake toward 5 g of salt or 87 mmol of sodium per day. 53
We recommend that patients with ADPKD who are <50 years old with eGFR >60 mL/min/1.73 m2 and without significant cardiovascular comorbidities should have a target BP of ⩽110/75 mm Hg, realizing that in some patients an individual target may be needed.
No change. We recommend salt restriction be followed as per current guidelines published by Hypertension Canada.
ADPKD-Specific Treatment Options
The Replicating Evidence of Preserved Renal Function: an Investigation of Tolvaptan Safety and Efficacy in ADPKD (REPRISE) trial was a phase 3 study designed to assess the efficacy and safety of tolvaptan in patients with advanced ADPKD using more frequent monitoring for liver toxicity. 54 The trial included patients 18 to 55 years of age with eGFR 25 to 65 mL/min/1.73 m2 or 56 to 65 years of age with eGFR of 25 to 44 mL/min/1.73 m2. The trial comprised a 1- to 2-week screening phase, a 1-week placebo run-in phase, a 2-week tolvaptan titration phase, and a 3-week tolvaptan run-in phase before patients were randomized to treatment with tolvaptan or placebo for 12 months. Follow-up was carried out 2 weeks after the end of the 12-month period. The primary endpoint of the study was change in eGFR from baseline to follow-up, with adjustment for the duration each patient was in the trial, interpolated to 1 year. The decline in eGFR from baseline to follow-up was significantly slower in the tolvaptan group (−2.34 mL/min/1.73 m2) compared with the placebo group (−3.61 mL/min/1.73 m2; P < .001), demonstrating the efficacy of tolvaptan in this patient population. Elevated alanine aminotransferase (ALT) levels >3 times the upper limit of normal (ULN) were observed in 5.6% of patients treated with tolvaptan compared with 1.2% of patients in the placebo group. Levels returned to normal following discontinuation of tolvaptan.
Following the results of these two phase 3 trials, that is TEMPO 3:4 and REPRISE, the Food and Drug Administration (FDA) recently approved tolvaptan to slow disease progression of ADPKD in adults. Similar to the Canadian approbation, tolvaptan is available in the United States through a restricted distribution program because of the risk of serious liver injury including, unfortunately, the need for a liver transplant in a treated patient from Japan.
The results of a phase 2 study comparing the tyrosine kinase inhibitor bosutinib to placebo in patients with ADPKD showed that bosutinib administered at 200 mg/day reduced kidney growth 7 ; however, no change in eGFR was observed. The overall gastrointestinal and liver toxicity profile was consistent with the profile in prior studies of bosutinib, with no new toxicities identified.
A post hoc analysis of the HALT-PKD study did not show differences in outcomes (percentage change in TKV for study A; composite of time to ESRD, death, or 50% reduction in eGFR for study B; and change in eGFR for both studies) between patients who never used statins and patients who used statins for at least 3 years. 55 Although this study did not demonstrate a benefit of statin therapy, it may be considered in cases where there are other indications for chronic kidney disease (CKD). Further studies are required to evaluate the benefits of statin therapy in patients with ADPKD. 56
An interim analysis of the Developing Interventions to Halt Progression of ADPKD 1 (DIPAK-1) study showed an increased risk of hepatic cyst infection with lanreotide treatment compared with standard care in female patients with ADPKD. 57 Based on these results, comanagement of large hepatic cysts with a hepatologist in female patients with ADPKD should be considered. This study is ongoing and final data regarding the risk-benefit ratio of lanreotide are pending. For guidance on the management of renal complications including cyst infection, nephrolithiasis, hematuria, and chronic pain, we refer the reader to treatment paradigms published by Lanktree and Chapman. 9
We suggest that all patients be referred to a nephrologist for initial assessment to determine what treatment should be initiated, in particular to initiate tolvaptan as soon as possible in patients determined to be appropriate candidates who would benefit from this therapy. We recommend treatment with tolvaptan for patients who fulfill the enrollment criteria of the TEMPO 3:4 study: 18 to 50 years of age, Cockcroft-Gault GFR >60 mL/min, and TKV >750 mL. In the absence of Cockcroft-Gault GFR, CKD-EPI >45 mL/min may be used, and in the absence of TKV, US KL >16.5 cm may be used. We suggest treatment with tolvaptan for patients who, according to the Mayo Clinic Classification, are classified as 1D or 1E with eGFR in CKD stage 3 or higher. Treatment with tolvaptan should be considered for patients who are classified as 1C and are <50 years old or have other risk factors for rapid progression. We do not recommend tolvaptan for patients classified as 1A or 1B. We suggest that treatment with tolvaptan be stopped when the patient develops ESRD. In the predialysis setting, there are no data to guide when treatment with tolvaptan should be stopped.
We recommend considering treatment with tolvaptan for patients who fulfill the enrollment criteria of the TEMPO 3:4 study: 18 to 50 years of age with TKV >750 mL and eGFR >45 mL/min/1.73 m2. We recommend treatment with tolvaptan for patients who fulfill the enrollment criteria of the REPRISE study: • 18 to 55 years of age with eGFR of 25 to 65 mL/min/1.73 m2 OR • 56 to 65 years of age with eGFR of 25 to 44 mL/min/1.73 m2 with historical evidence of a decline in eGFR >2.0 mL/min/1.73 m2/year. We believe that, although there were no inclusion criteria for kidney size, based on the abundance of evidence that increased size of kidneys is relevant, these REPRISE criteria relate to those patients with ADPKD who have enlarged kidneys. In those patients with advanced or rapidly progressive CKD without enlarged kidneys, an alternate diagnosis for CKD should be investigated. We suggest treatment with tolvaptan for patients who, according to the Mayo Clinic Classification, are classified as 1D or 1E with eGFR in CKD stages 1-4 (eGFR >25 mL/min). Treatment with tolvaptan may be considered for patients who are classified as 1C and are <50 years old or have other risk factors for rapid progression, such as an annual decrease in eGFR of >2.5 mL/min/1.73 m2 and/or increase in TKV of >5% per year. We suggest that treatment with tolvaptan be stopped when the patient develops ESRD. In the predialysis setting with eGFR <25 mL/min/1.73 m2, there are no data to guide when treatment with tolvaptan should be stopped.
Additional Considerations When Giving Tolvaptan
Several studies were identified that provided additional information to consider when giving tolvaptan to patients with ADPKD. A post hoc analysis of the TEMPO 3:4 study showed that lower fasting morning urine osmolality, and therefore better eGFR, at baseline resulted in greater reduction in urine osmolality over 36 months of treatment with tolvaptan. 58 Patients with greater reduction in urine osmolality experienced significant reduction in decline in renal function.
A second post hoc analysis of the TEMPO 3:4 trial evaluated aquaretic adverse events (AAEs) attributable to excess free water clearance in patients with ADPKD treated with tolvaptan. 59 The study showed that AAEs were common but well tolerated, and that patients in earlier stages of disease progression were more sensitive to AAEs. After 3 years of treatment, 75% of patients in the tolvaptan arm reported that that they would be able to tolerate their current dose of tolvaptan for the remainder of their lives compared with 85% of patients in the placebo group. These results may be used to inform decisions regarding the dose and titration of tolvaptan in patients in earlier stages of disease progression so as to minimize AAEs and treatment discontinuation.
The open-label, 2-year extension trial TEMPO 4:4 was designed to assess the long-term efficacy and safety of tolvaptan in patients completing the TEMPO 3:4 study, and to compare slopes of TKV growth in early-treatment (former tolvaptan arm of TEMPO 3:4 study) versus delayed-treatment (former placebo arm of TEMPO 3:4 study) groups. 60 The change in TKV from baseline in TEMPO 3:4 to study end in TEMPO 4:4 was 29.9% for early-treatment patients and 31.6% in delayed-treatment patients. This difference was not significant, although the slope of TKV growth during TEMPO 4:4 was significantly higher in early-treatment patients (6.16%/year) compared with delayed-treatment patients (4.96%/year, P = .05). In addition, there was no difference in eGFR decline between early-treatment patients and late-treatment patients. Treatment with tolvaptan maintained the slowing of eGFR decline for an additional 2 years in TEMPO 4:4 (3.15 mL/min/1.73 m2, P < .001). The adverse event profile was similar between TEMPO 3:4 and TEMPO 4:4. The results of this study identified several key considerations when giving tolvaptan: (1) tolvaptan offers long-term benefit with a tolerable safety profile out to 5 years; (2) tolvaptan offers benefit to patients not only when initiated early but also when initiated later in the disease process; and (3) once patients are initiated on tolvaptan, slope of TKV growth and eGFR may be used to follow disease progression.
The TEMPO 3:4 extension Japan trial evaluated the long-term safety profile of tolvaptan up to an additional 3 years in patients with ADPKD. 61 The results of the study showed that extended use of tolvaptan did not increase the risk of hepatic or other adverse events, thus supporting the findings of the TEMPO 4:4 study. In this study, liver-related AEs were monitored monthly; therefore, this study validates the current paradigm that monthly monitoring for hepatic AEs is sufficient to identify early hepatocellular injury and to prevent those patients from reaching drug-induced liver injury (defined as 3 times the ULN in serum ALT or aspartate aminotransferase [AST] levels and 2 times the ULN in serum total bilirubin).
We recommend that patients with ADPKD on treatment with tolvaptan follow a sodium-restricted diet of ⩽2.4 g/day (⩽100 mmol/day). We suggest titrating tolvaptan to the maximal tolerated dose or to achieve a uOSM <250 mOsm/kg water. Consideration should be given to consultation with a dietician to minimize sodium and osmolal intake to help manage severe AAEs.
Footnotes
Appendix
Summary of Updated Recommendations.

| Identifying patients with ADPKD 1. We suggest that all patients with a diagnosis of ADPKD or suspected ADPKD be referred to a nephrologist for initial assessment. Initial assessment should include kidney imaging and, in some cases, genetic testing to determine the patient’s risk of rapid progression and to determine what treatment should be initiated. |
| Genetic testing 1. Based on existing data, genetic testing is not necessary for selecting treatment options for patients with a confirmed diagnosis of ADPKD. |
| Renal imaging for diagnosis 1. The preferred method for confirming the presence of ADPKD in patients with a family history is US imaging and the use of the Unified Criteria to establish diagnosis and determine if typical or atypical. 2. In select circumstances, such as in patients without a family history of ADPKD, other imaging modalities, including CT or MRI, may be considered to diagnose ADPKD, particularly to detect cysts in younger patients. |
| Renal imaging for prognosis and disease progression 1. We recommend that a baseline assessment of renal size be undertaken in patients with ADPKD. The objective of these measurements is to determine which patients are at risk of rapid progression and may be suitable candidates to be considered for therapeutic intervention based on their risk of progression. 2. In patients with typical morphology, we recommend using US to measure KL, and MRI or CT to measure TKV (and to calculate htTKV where appropriate) or if a more precise measurement is required for therapeutic decisions. In cases where historical images are available, those images should be consulted before requesting new imaging. 3. After a baseline assessment of renal size, not all patients require routine reassessment of renal size. If renal size reassessment is performed, we recommend it should not exceed a frequency of once yearly. |
| Assessing disease progression 1. We recommend that in current clinical practice, patients with an htTKV measurement be categorized in terms of their risk of progression as per the Mayo Clinic Classification or other validated clinical tools. 2. Currently available TKV-based prognostication tools should be applied only to class 1 (typical morphology) patients, as we suggest that these patients are likely to be rapid progressors. Certain patients may require further clinical evaluation. |
| 3. We suggest that patients be considered at risk of rapid progression of ADPKD renal disease if they meet either of the following criteria: (1) classified as Mayo class 1C, D, or E, or (2) have an US KL of >16.5 cm bilaterally. 4. We suggest the following be used as markers of rapid progression: (1) a sequential increase of >5% annually in htTKV on imaging, or (2) documented disease progression (eg, rapid decline in eGFR, defined as decline in eGFR >2.5 mL/min/1.73 m2; patients in the placebo group of the TEMPO 3:4 study showed a decline in eGFR of 3.5 mL/min/1.73 m2). |
| Nontargeted treatment options 1. We recommend that patients with ADPKD who are <50 years old with eGFR >60 mL/min/1.73 m2 and without significant cardiovascular morbidities should have a target BP of ⩽110/75 mm Hg, realizing that in some patients an individual target may be needed. 2. We recommend salt restriction be followed as per current guidelines published by Hypertension Canada. |
| ADPKD-specific treatment options 1. We recommend treatment with tolvaptan for patients who fulfill the enrollment criteria of the TEMPO 3:4 study: 18 to 50 years of age with TKV >750 mL and eGFR >45 mL/min/1.73 m2. 2. We recommend treatment with tolvaptan for patients who fulfill the enrollment criteria of the REPRISE study: • 18 to 55 years of age with eGFR 25 to 65 mL/min/1.73 m2 OR • 56 to 65 years of age with eGFR of 25 to 44 mL/min/1.73 m2 with historical evidence of a decline in eGFR >2.0 mL/min/1.73 m2/year We believe that, although there were no inclusion criteria for kidney size, based on the abundance of evidence that increased size of kidneys is relevant, these REPRISE criteria relate to those patients with ADPKD who have enlarged kidneys. In those patients who do not, an alternate diagnosis for CKD should be investigated. 1. We suggest treatment with tolvaptan for patients who, according to the Mayo Clinic Classification, are classified as 1D or 1E with eGFR in CKD stages 1-4 (eGFR >25 mL/min). Treatment with tolvaptan may be considered for patients who are classified as 1C and are <50 years old or have other risk factors for rapid progression, such as an annual decrease in eGFR of >2.5 mL/min/1.73 m2 and/or increase in TKV of >5% per year. 2. We suggest that treatment with tolvaptan be stopped when the patient develops ESRD. In the predialysis setting with eGFR <25 mL/min/1.73 m2, there are no data to guide when treatment with tolvaptan should be stopped. |
| Additional considerations when giving tolvaptan 1. We recommend that patients with ADPKD on treatment with tolvaptan follow a sodium-restricted diet of ⩽2.4 g/day (⩽100 mmol/day). 2. We suggest titrating tolvaptan to the maximal tolerated dose or to achieve a uOSM <250 mOsm/kg water. Consideration should be given to consultation with a dietician to minimize sodium and osmolal intake to help manage severe AAEs. |
Note. ADPKD = autosomal dominant polycystic kidney disease; US = ultrasound; CT = computed tomography; MRI = magnetic resonance imaging; KL = kidney length; TKV = total kidney volume; htTKV = height-adjusted total kidney volume; eGFR = estimated glomerular filtration rate; TEMPO = Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes; BP = blood pressure; REPRISE = Replicating Evidence of Preserved Renal Function: an Investigation of Tolvaptan Safety and Efficacy in ADPKD; ESRD = end-stage renal disease; AAEs = aquaretic adverse events.
Acknowledgements
The authors gratefully acknowledge the contribution of Angela Styhler in the drafting of the article.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Steven Soroka reports receiving honoraria for lecturing on autosomal dominant polycystic kidney disease (ADPKD), and developing educational material and participating in advisory boards from Otsuka Canada. Dr Daniel G. Bichet reports receiving honoraria for lectures on ADPKD and grants from Otsuka Canada, grants from Otsuka US to conduct trials, and Otsuka Europe for lectures and consultancies on ADPKD. Dr Ahsan Alam reports receiving honoraria for consultancy and lecturing from Otsuka Canada and Amgen. Dr Louis-Philippe Girard reports receiving honoraria for his involvement in continuing medical education and his participation in advisory boards from Otsuka Canada. Dr Philip McFarlane reports receiving honoraria for his participation in advisory boards from Otsuka Canada. Dr Paul Tam reports receiving a research grant from Janssen and honoraria for his participation in advisory boards from Amgen. Dr. Sanjaya Pandeya reports receiving honoraria for participation in advisory boards, consultancy, and lecturing from Otsuka Canada. Dr Micheli Bevilacqua discloses that he has received honoraria for consultancy, lecturing, and participation in advisory boards, as well as a research grant from Otsuka Canada. Drs Paul Komenda and Rolf Loertscher have no disclosures to report regarding their contributions to this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Otsuka, the funding sponsor, offered unrestricted support to the development of these recommendations and did not have any part in creating this document. Funding sponsor representatives were not present at the meeting. The meeting that produced the recommendations presented was organized by SNELL Medical Communication. Honoraria were provided to the participants to review select articles and attend a meeting to discuss and provide expert opinion on the topics contained herein. The funding also provided the authors with the services of an experienced and qualified medical writer to ensure a professional article. The medical writer, solely under the direction and outline of the authors, assisted in researching the topic and preparing the article draft.