
Abstract
Progressive multifocal leukoencephalopathy (PML) is a rare disease of the central nervous system caused by opportunistic infection with JC virus. It presents in patients who are immunocompromised, and diagnosis is made by correlating clinical findings and radiological changes with the detection of JC virus in cerebrospinal fluid. Rarely, a brain biopsy is needed. A 72 year old with high grade B-cell lymphoma developed right arm weakness and limb ataxia shortly after his diagnosis. CNS involvement was excluded with a normal CT head, MRI brain/spine, and CSF examination. A paraneoplastic cause was suspected, and he received 5 cycles of Rituximab-containing chemotherapy to a complete metabolic remission. His neurology evolved during treatment despite serial MRI and CSF examination remaining normal. CSF and serum were both negative for JC virus by PCR. Following completion of chemotherapy, he deteriorated acutely with seizures and personality changes. It was only at this point that a repeat MRI showed new multiple scattered ring enhancing lesions within both cerebral hemispheres. The patient underwent a brain biopsy confirming JC virus positive-PML by immunohistochemistry and passed away one month later. This case illustrates the diagnostic challenges associated with PML and had several atypical features which led to diagnostic delay, specifically the onset of symptoms before starting immunochemotherapy, and the lack of radiological change despite evolving neurology. The eventual MRI abnormalities were not altogether classical for PML which, coupled with the JC-negativity in CSF and serum, meant a brain biopsy was required to reach the diagnosis.
Keywords
Case
A previously fit and well, 72-year-old man was diagnosed with Stage 4A diffuse large B-cell lymphoma after an incidental finding of asymptomatic lymphadenopathy. Shortly after diagnosis he developed right arm weakness, limb ataxia and myoclonic jerks. A normal MRI brain/spine and cerebrospinal fluid (CSF) examination excluded central nervous system involvement with lymphoma. A paraneoplastic syndrome was suspected and the patient received 5 cycles of rituximab-containing chemotherapy to complete remission.
After 4 cycles of chemotherapy, he reported new left arm weakness. A formal neurology review documented mild global weakness (4/5 power) and areflexia, significant limb ataxia, and choreatic movements. Two further MRI brain/spine showed no new abnormality. Repeat CSF examination remained normal including a negative PCR virology screen (JCV DNA and intrathecal JCV antibody not detected). Serology for anti-neuronal antibodies, Voltage-Gated-Potassium-Channel antibodies and N-methyl-D-aspartate antibodies was negative. LDH and CRP were unremarkable. A paraneoplastic syndrome remained a consideration although the choreatic movements were atypical. Tetrabenazine was commenced for symptomatic relief of hyperkinesis.
One month later, he deteriorated acutely with seizures and personality change. CT brain was normal but MRI now demonstrated bilateral open ring enhancing lesions, particularly within the right parietal lobe with surrounding oedema (Figure 1) and abnormal meningeal enhancement within the right occipital lobe. An infective/inflammatory cause was thought likely given the rapid clinical deterioration and diffuse radiological appearance. Chemotherapy was stopped. Two further lumbar punctures confirmed a newly-elevated CSF protein (637 mg/L). CSF remained PCR-negative for viruses (mumps, EBV, CMV, VZV, HSV, enterovirus, parenchovirus), and toxoplasmosis. Gram-stain and culture for tuberculosis was negative. Serology for Lyme disease, streptococcus, toxoplasmosis, syphilis, HIV, hepatitis B & C and fungus was also negative.
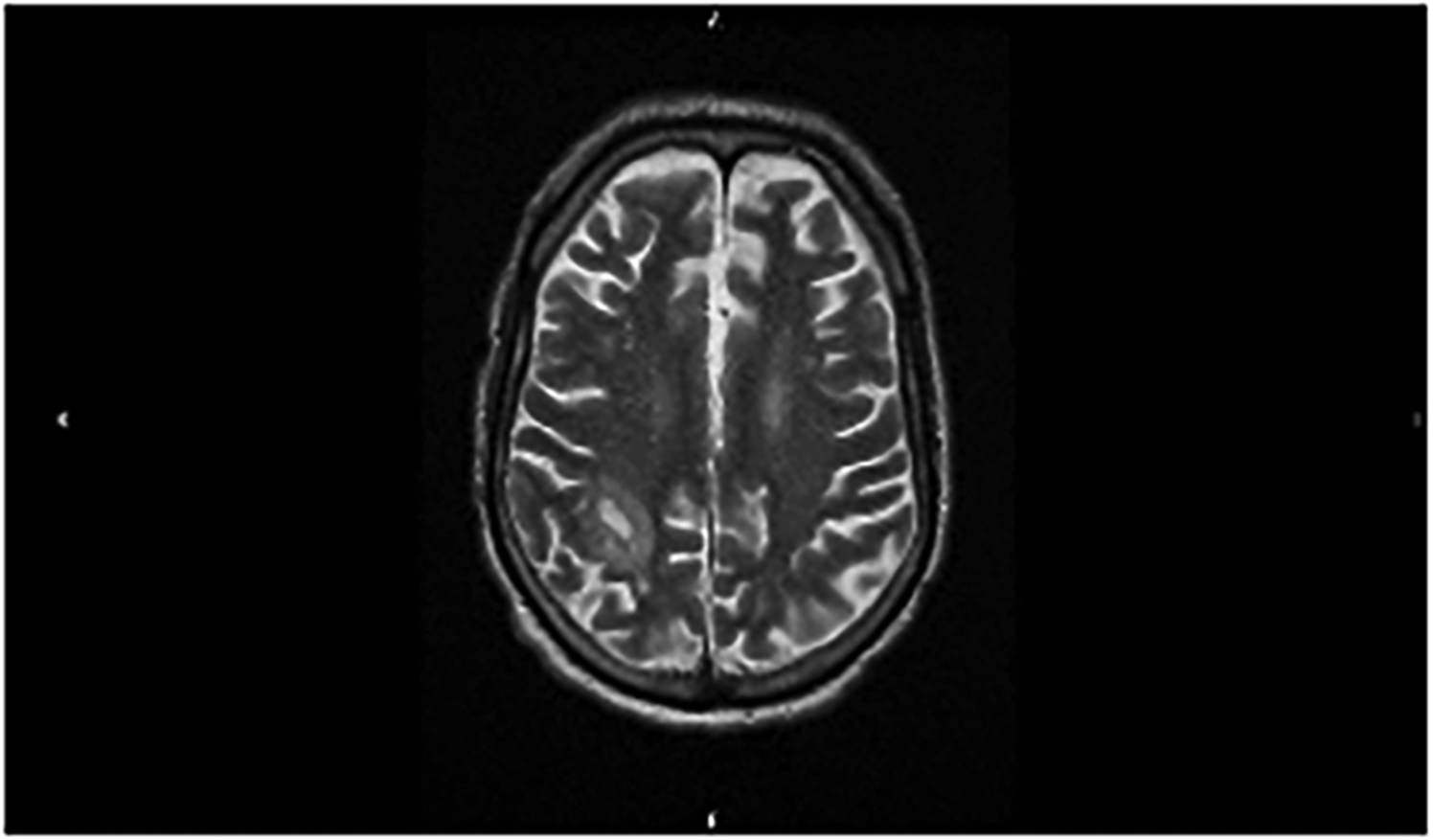
T2-weighted MRI image showing open ring enhancement in right parietal lobe. 49 × 29 mm (150 ×150 DPI).
The patient underwent a craniotomy and biopsy of the intra-cranial lesions. Histology reported large areas of demyelination with relative preservation of axons, and extensive infiltration by small lymphocytes, foamy macrophages, and reactive astrocytes (Figure 2a). Numerous enlarged oligodendroglial cells containing pale intranuclear inclusions stained positively with JCV antibodies (SV40 and P53) (Figure 2b). The appearance was characteristic of PML. In view of poor performance status and lack of clinically-proven treatments, the patient was palliated and died 1 month later. Time from initial presentation to PML diagnosis was 6 months.
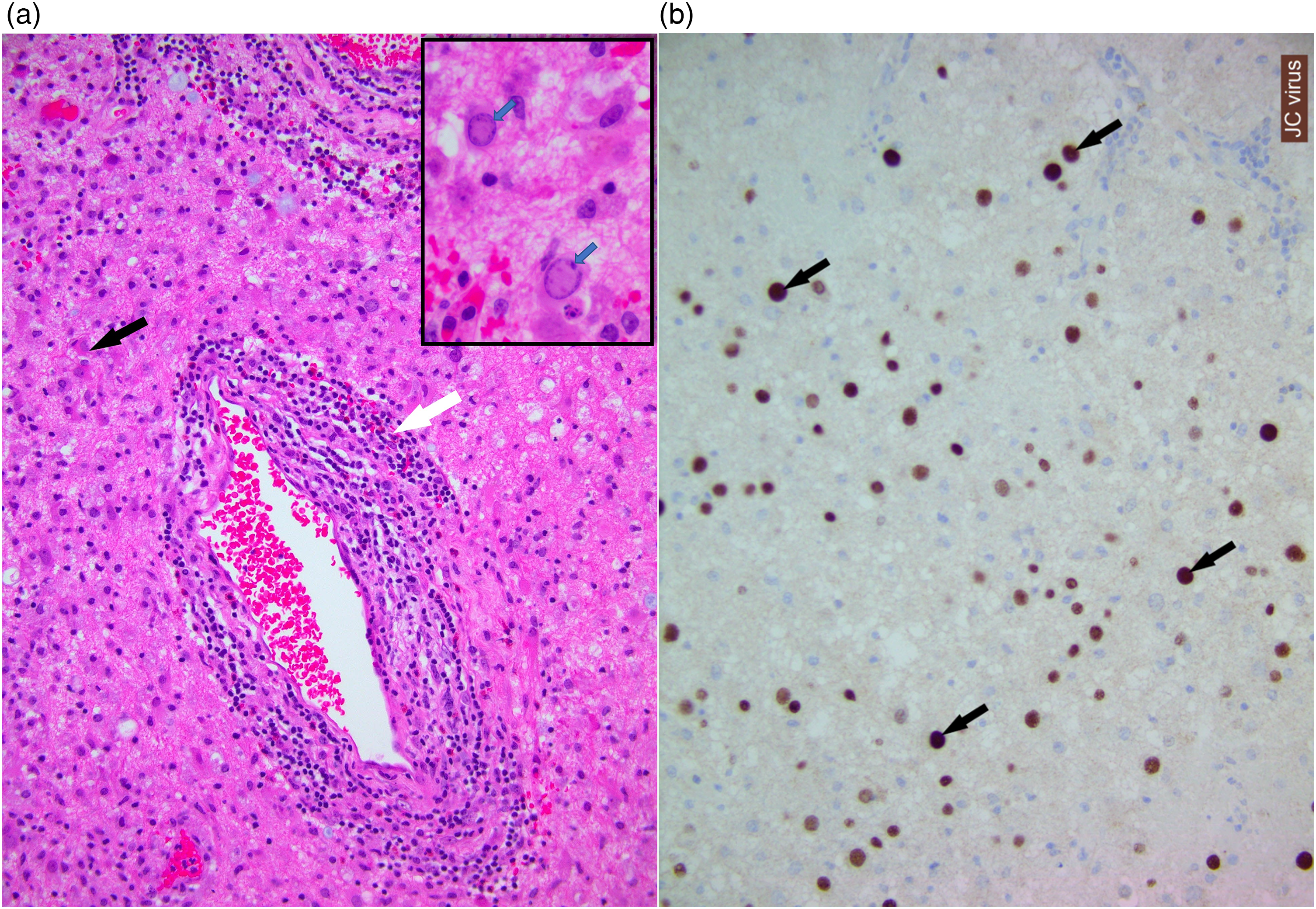
(a) Histology from brain (HE stain, x200) showing chronic inflammatory cells in the parenchyma and around blood vessels (white arrow). There are scattered reactive astrocytes (black arrow) suggesting gliosis. Inset shows rounded enlarged oligodendroglial cells with pale intranuclear viral inclusion (blue arrows). (b) Immunohistochemistry using SV40 (JC virus) antibody highlights viral inclusions (brown staining) within oligodendroglial cells (black arrows). (x200).
Discussion
PML is an opportunistic infection caused by John Cunningham virus (JCV), and is usually fatal. 1 Antibodies against JCV are detected in 50–90% of adults, and latent virus is harboured within kidneys, marrow and lymphoid tissue. 2 In immunocompromised people, viral reactivation enables infection of glial and neuronal cells causing oligodendrocytic demyelination. 2 This is particularly associated with haematological malignancy, HIV/AIDS, and immunosuppressive treatments including anti-CD20 monoclonal antibodies (eg. rituximab).2,3
Symptoms of PML include cognitive/behavioural disturbance, sensorimotor deficit, gait abnormality, incoordination, and visual/speech disturbance. Headaches and seizures are less common.1,3 Radiologically, MRI is the preferred diagnostic tool due to higher sensitivity over CT. 4 MRI lesions are typically ill-defined, often multi-focal, and non-contrast enhancing. 5 Once PML is suspected, CSF analysis for JCV detection should be performed. PCR was first used to detect JCV in 1989 and is now the gold standard with 75% sensitivity and 96% specificity. 6 Ultrasensitive PCR (95% sensitivity) has been developed but is not widely available. 6 Diagnostic yield can be improved by serial CSF analysis as JCV-negative cases can become positive as lesion volume increases. 7 When JCV remains undetectable in CSF, brain biopsy may be required.1,8 Characteristic histology comprises demyelination, enlarged oligodendroglial nuclei, and bizarre astrocytes. Immunohistochemistry and PCR can be applied to brain tissue to detect JCV antigens and DNA respectively. 8
Our patient had some unusual features worthy of discussion. Firstly, his neurological symptoms were almost synchronous with lymphoma diagnosis and prior to any rituximab-containing chemotherapy. He later developed choreatic movements which are unusual in PML. 1 PML is a well-recognised complication of haematological malignancies, affecting 0.07% overall with the highest risk (0.5%) in chronic lymphocytic leukaemia (CLL). 9 13% of all PML cases are associated with haematological malignancy 9 and, in contrast to our case, most occur following treatment. In a study of 16 patients with haematological malignancies, 15 had chemotherapy and 9 underwent stem cell transplantation prior to PML diagnosis. The median time from cancer diagnosis to PML diagnosis was 48.5 months. 2 In a case series of 57 PML patients who had received rituximab (52 had B-cell lymphoproliferative disorders), 65% were diagnosed within the first 2 years of treatment. The median time from first rituximab to PML diagnosis was 16 months. 9 Signs of PML at the time of lymphoma diagnosis with no prior rituximab/chemotherapy is rare.
Secondly, there was absence of radiological change despite evolving neurology. The high sensitivity of MRI allows the detection of subtle PML-related changes several months before symptoms manifest.4,5 A study of 372 PML cases found 30 (8.1%) were asymptomatic when lesions were first detected on MRI. Median time from lesion detection to PML confirmation was 12 days (range 0–168). 4 Our patient's neurological symptoms preceded any MRI abnormality by 4 months. MRI changes only became apparent after acute deterioration with seizure activity. Furthermore, there was a mixed radiological picture with some features strongly suggestive of PML (juxta-cortical positioning of lesions, restricted diffusion at leading edge) whilst others were atypical (contrast enhancement). This added yet more challenge to the diagnostic complexity.
Treatments for non-HIV-associated PML are ineffective. In a series of 57 patients with rituximab-associated PML, a 90% fatality rate was reported with median survival from diagnosis only 2 months. 9 Similarly, in a series of 16 patients with haematological malignancy-associated PML, there was no consensus on optimal treatment and the median survival of patients who received PML-directed therapy was 4.3 months versus 0.87 months for those who did not. 2 Recently, the PD-1 inhibitor Pembrolizumab has shown promise in a small number of PML patients (5/8 improved clinically alongside a reduction in JCV load) 10 but further evidence is needed.
In conclusion, PML should be considered in any immunocompromised patient with unexplained neurology irrespective of the timing of onset. Serial MRI and CSF analysis may increase diagnostic yield but clinicians need to recognise the limitations of currently-available PCR for JCV detection in CSF. Brain biopsy should be considered in uncertain cases.
Footnotes
Contributor statement
We would like to acknowledge the following colleagues for their contribution to our project:
Dr Anthony George, Consultant Neuroradiologist at Torbay & South Devon NHS Foundation Trust who interpreted the MRI brain image.
Dr Aditya Shivane, Consultant Neuropathologist at University Hospitals Plymouth NHS Trust who provided and interpreted the histology slides.
Competing Interests
None declared.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Statement regarding patient consent
Permission sought from patient's wife who has given written consent for us to publish this case (patient is deceased).