
Abstract
Acute liver failure is a life-threatening condition commonly caused by drug-induced hepatotoxicity or viral hepatitides. However, there are a number of rarer causes such as haemophagocytic lymphohistiocytosis. Haemophagocytic lymphohistiocytosis is a syndrome of uncontrolled immune cell activation, triggered by infection or malignancy, which carries a high mortality. Whilst mild to moderate liver injury is commonly seen with haemophagocytic lymphohistiocytosis, acute liver failure has rarely been reported in adults. We present a case of a 74-year-old man with acute liver failure secondary to haemophagocytic lymphohistiocytosis triggered by undiagnosed large B-cell lymphoma. Initially treated for biliary sepsis, there was a delay in the diagnosis of haemophagocytic lymphohistiocytosis and despite initiating chemotherapy, he died soon after. This case highlights the importance of considering haemophagocytic lymphohistiocytosis as a rare cause of acute liver failure, as given the life-threatening potential of haemophagocytic lymphohistiocytosis, a prompt diagnosis may allow early initiation of chemotherapy for any chance of survival.
Keywords
Background
Acute liver failure (ALF) is a rare condition that varies in aetiology and prognosis. Paracetamol-induced hepatotoxicity in the United Kingdom and viral hepatitis globally are the predominant causes; however, a significant number of cases remain undetermined.1,2 Haemophagocytic lymphohistiocytosis (HLH), a syndrome of uncontrolled immune cell activation, can occur as a primary or acquired disorder. 3 Mild to moderate liver injury is a common complication of Haemophagocytic lymphohistiocytosis (HLH) in 80% of patients. 4 However rarely, patients may present with Acute Liver Failure. 5 The mortality of Haemophagocytic lymphohistiocytosis ranges between 41% and 75% and with associated Acute Liver Failure the prognosis is significantly poorer.4,6 Due to the rarity of the Acute Liver Failure caused by Haemophagocytic lymphohistiocytosis, the diagnosis is often not considered, resulting in delayed treatment and contributing to the high mortality. Herein we present a rare case of an elderly patient who developed Acute Liver Failure secondary to Haemophagocytic lymphohistiocytosi caused by an underlying newly diagnosed B-cell lymphoma, a review of the literature and lessons learnt.
Case presentation
A 74-year-old male presented with a one-month history of worsening pruritus, confusion with multiple falls and fever. He had suffered with lethargy and night sweats for a number of months. There was no history of liver disease but had had previous coronary stenting for ischaemic heart disease for which he was taking aspirin. He was a non-smoker and occasionally consumed alcohol.
On arrival, he was pyrexial and tachycardic but abdominal examination was unremarkable. Initial blood tests revealed: deranged liver function (Bilirubin 28, Alanine transaminase 106, Alkaline phosphatase 600) with synthetic impairment (INR 2.0, Albumin 29), pancytopenia (Haemoglobin 85; White Cell Count 2.7, Platelets 71) and raised C-reactive protein 55 (Figures 1 and 2). On the basis of a fever and deranged liver function, the patient was initiated on co-amoxiclav for biliary sepsis. However, an abdominal ultrasound showed a thickened gallbladder without any stones nor biliary duct dilatation, and a normal liver and portal venous flow. The spleen was enlarged at 16 cm containing a hypoechoic area 30 × 43 mm. The latter finding was confirmed by a CT abdomen (Figure 3). A subsequent magnetic resonance cholangiopancreatography was normal. The work-up for acute liver injury including viral serology, autoimmune screen and immunoglobulins was unrevealing. Several blood cultures taken for ongoing spiking temperature were negative.
Full blood count and clotting results. Liver function results. CT image demonstrating an enlarged spleen at 16 cm.


Differential diagnosis
The differential diagnosis included a splenic abscess and haematological malignancy given the imaging so he was discussed in the haematology cancer multidisciplinary meeting.
Clinical course
Despite initial treatment with co-amoxiclav, he continued to spike fevers and suffered haemodynamic instability necessitating two medical emergency calls. Antibiotics were escalated to Piperacillin/Tazobactam on Day 4. Blood tests showed raised lactate, worsening pancytopenia, acute kidney injury and further deterioration in liver function with the development of jaundice and INR up to 2.0 (Figures 2 and 4). Initial Lactate dehydrogenase was raised at >2000 IU/L and ferritin >3000 ug/L.
Urea and electrolyte results.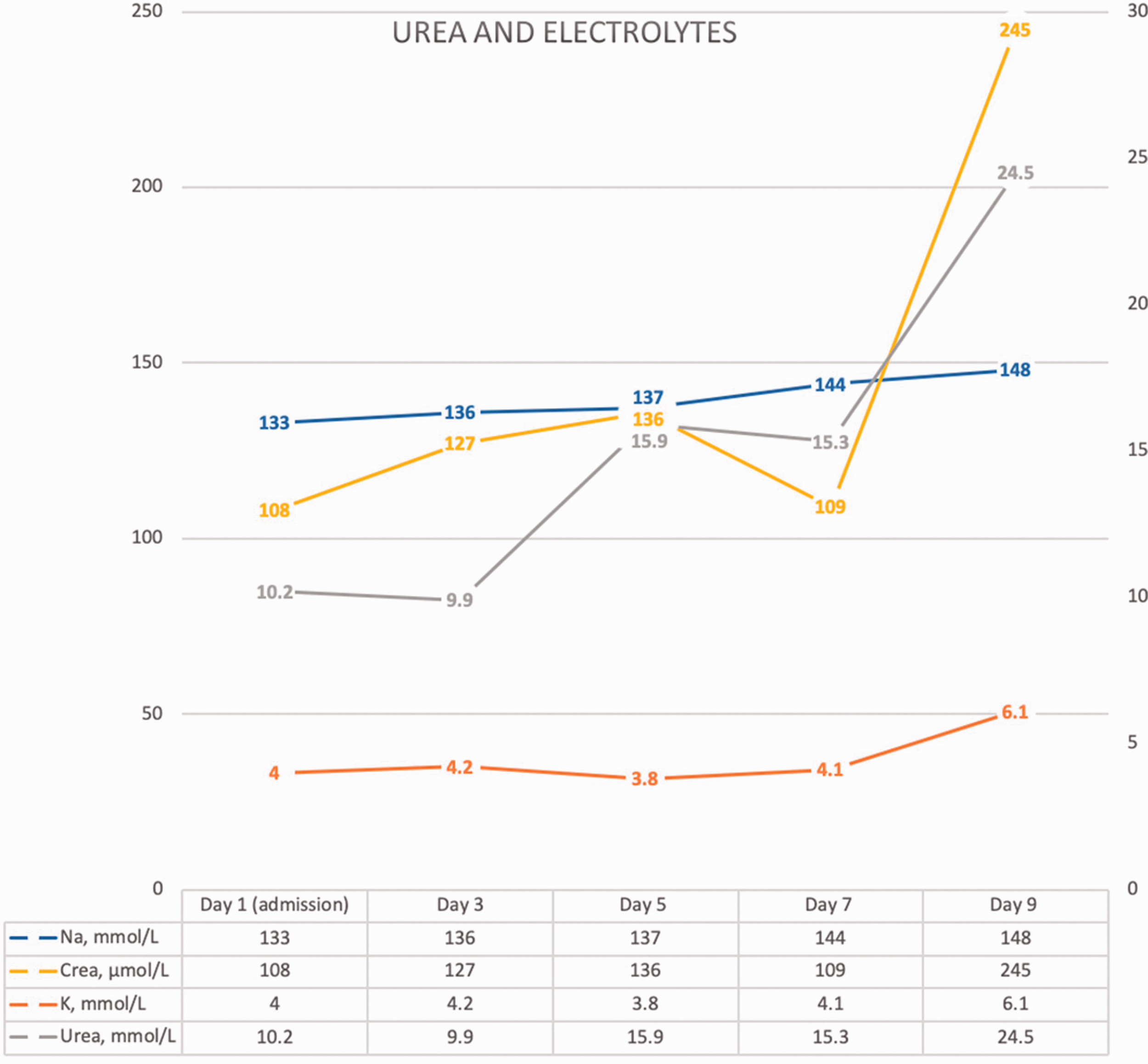
He had a staging CT as per haematology multidisciplinary team recommendation, which showed new bilateral pleural effusions, minimal ascites and peri-pancreatic stranding suggestive of mild pancreatitis. However, the clinical picture was not in keeping with acute pancreatitis and the amylase was only marginally raised at 125 iu/L, so this was deemed unlikely. The haematology team performed a bone marrow aspirate and trephine biopsy. The patient further deteriorated with development of hepatic encephalopathy, hypoxia and hypotensive with a persistent tachycardia. The patient was given fluid resuscitation, and the antibiotics were further escalated to Meropenem. A hepatology review recommended to check for Hepatitis A, Hepatitis E, Hepatitis B core antibody, leptospirosis, Brucella, and herpes simplex virus serology, all of which were normal.
HLH-2004 diagnostic criteria fulfilled.21

Bone marrow trephine biopsy result eventually confirmed diffuse large B-cell lymphoma with 80% blast, confirming haemophagocytic lymphohistiocytosis with underlying malignancy (Figure 5).
Bone marrow trephine biopsy images. (a) Microscopy showing atypical cells with irregular nuclei and prominent nucleoli (×600). (b) Immunohistochemistry to show B cell nature of atypical cells (×400).
Outcome and follow-up
The patient died as an inpatient during his admission.
Discussion
Haemophagocytic lymphohistiocytosis is a clinical syndrome of excessive macrophage activation 7 which rarely causes acute liver failure. Herein we have presented a case of acute liver failure secondary to haemophagocytic lymphohistiocytosis due to an undiagnosed large B-cell lymphoma in an elderly gentleman. haemophagocytic lymphohistiocytosis is classified as primary or secondary, resulting in defective natural killer cell function. The primary form, usually seen in infants and children, results from genetic missense mutations in the perforin genes, responsible for NK cell and cytotoxic T lymphocyte function.8,9 Secondary haemophagocytic lymphohistiocytosis, seen in adults, is caused by infections, malignancy, rheumatological and metabolic diseases. Epstein–Barr virus is the most consistent, in up to a third of secondary haemophagocytic lymphohistiocytosis. 10 Malignancies are accountable for up to 27% of secondary haemophagocytic lymphohistiocytosis and of these haematological are of the highest prevalence. 11
Malignancy-associated haemophagocytic lymphohistiocytosis
Previously a rare entity, malignancy-associated haemophagocytic lymphohistiocytosis has been shown in recent retrospective studies to be evident in up to 1% of underlying malignancies at diagnosis. For rare subtypes of B-cell lymphoma, it could be as high as 20% (intravascular B-cell lymphoma or B-cell lymphoma without peripheral adenopathies). 12 Malignancy associated haemophagocytic lymphohistiocytosis may present as a feature of an undiagnosed malignancy as in our patient with B-cell lymphoma, or upon initiation of immune modulating therapies. It is understood that the disruption of immune homeostasis from malignancies and associated therapies promoting a degree of T-cell dysfunction can act as the trigger for Malignancy associated haemophagocytic lymphohistiocytosis. 13
Liver injury in haemophagocytic lymphohistiocytosis
Liver injury is a common complication of haemophagocytic lymphohistiocytosis with varying degrees of insult but acute liver failure is rarely reported. The pathophysiology of acute liver failure in haemophagocytic lymphohistiocytosis remains largely unknown. It is theorised that infiltration of activated haemophagocytic histiocytes or the increased cytokine production associated with haemophagocytic lymphohistiocytosis results in liver injury. 14 This can be further compounded by liver injury caused by the underlying disease.15,16 The histopathological findings in the liver of patients with haemophagocytic lymphohistiocytosis are non-specific and can include hepatocellular necrosis, sinusoidal dilatation, endothelialitis and steatosis. 3
B-cell lymphoma can present rarely as acute liver failure 17 and this is usually as a manifestation of end-stage malignant disease. 18 The liver injury and extensive cholangitis are due to malignant infiltrates of intrahepatic ducts, hepatic venules and the hepatic parenchyma itself. 19 In our case, imaging failed to demonstrate any liver infiltrates and the liver appeared normal, therefore suggesting haemophagocytic lymphohistiocytosis as the predominant cause of the acute liver failure rather than due to malignant infiltration.
Presentation and diagnosis of haemophagocytic lymphohistiocytosis
Diagnosis of haemophagocytic lymphohistiocytosis is challenging due to its rare occurrence and non-specific presentation. 9 Patients with haemophagocytic lymphohistiocytosis characteristically present with a varying picture of cytopenia, fever and multiple organ involvement. 20 The HLH-2004 study of paediatric patients devised criteria for diagnosis. The following clinical findings were common: Fever (95%), splenomegaly (89%), bicytopenia (92%), hypertriglyceridaemia or hypofibrinogenaemia (90%), haemophagocytosis (82%), ferritin >500 mcg/L (94%), low/absent NK cell activity (71%) and soluble CD25 elevation (97%).21,22 To fulfil the diagnostic criteria, five of the eight are required. Alternatively, it can be diagnosed with genetic testing. 21 These criteria are based on paediatric patients and there is an unmet need for adult specific criteria, in particularly for m-haemophagocytic lymphohistiocytosis, in order to prevent missed or delayed diagnosis in adults.
Our patient met five out of the eight criteria required for diagnosis of haemophagocytic lymphohistiocytosis per HLH-2004 (Table 1). One of the remaining criteria of haemophagocytosis in the bone marrow was not seen in our case – it showed reactive changes only. In fact, often haemophagocytosis may not be seen on the initial bone marrow as pathological changes due to haemophagocytic lymphohistiocytosis may take days or weeks to manifest and it is not considered pathognomonic for haemophagocytic lymphohistiocytosis. Thus, the absence of haemophagocytosis on the initial bone marrow should not delay treatment in cases where there is a high clinical suspicion for haemophagocytic lymphohistiocytosis. 13
The clinical course of Malignancy associated haemophagocytic lymphohistiocytosis is often rapidly progressing and characterised by poor outcomes. 13 The management of Malignancy associated haemophagocytic lymphohistiocytosis is a focus of debate. Whilst it seems logical to treat the underlying cause, these patients are in a hyperinflammatory state with an ongoing cytokine storm and giving chemotherapy to treat an underlying malignancy may have a deleterious effect. An approach that involves controlling the cytokine storm and the underlying malignancy would be preferable. 13
In our case, the underlying cause of haemophagocytic lymphohistiocytosis was unknown and we had no evidence to suggest that a malignant process was driving the haemophagocytic lymphohistiocytosis and the more common cause of viral infection was higher in the differential diagnosis. It was decided to initiate chemotherapy with methylprednisolone as the patient’s condition was deteriorating, in the hope that he may respond and with no other treatment options. Unfortunately, he most certainly developed tumour lysis syndrome due to undiagnosed B-cell lymphoma after giving methylprednisolone which hastened his death.
Conclusion
Our case emphasises the importance of considering haemophagocytic lymphohistiocytosis as a rare cause of acute liver failure. A condition more common in paediatrics, recent literature shows an increasing prevalence of haemophagocytic lymphohistiocytosis in adults with poor prognoses.23,24 A delay in diagnosis is a key contributor to the mortality rate,4,6,25 thus we hope to raise awareness amongst physicians about this diagnosis.
Patient consent
Written informed consent for patient information and images to be published was provided by the patient’s spouse.
Learning points/take home messages
Haemophagocytic lymphohistiocytosis is a rare cause of acute liver failure. A high clinical suspicion of haemophagocytic lymphohistiocytosis, even without haemophagocytosis on bone marrow, could prompt early treatment and possibly reduce mortality. A need for adult-specific diagnostic criteria for haemophagocytic lymphohistiocytosis which may lead to a prompt diagnosis. To be aware of and avoid diagnostic momentum – in this case, a previous diagnosis of biliary sepsis, without any radiological evidence and non-response to antibiotics.