LB01 EFFICACY AND SAFETY OF USTEKINUMAB FOR ULCERATIVE COLITIS THROUGH 2 YEARS: UNIFI LONG-TERM EXTENSION
B.E. Sands1, W.J. Sandborn2, R. Panaccione3, C. O’Brien4, H. Zhang4, J. Johanns4, Y. Zhou4, I. Tikhonov4, L. Peyrin-Biroulet5, G. Van Assche6, S. Danese7, S. Targan8, M.T. Abreu9, T. Hisamatsu10, E. Scherl11, R.W. Leong12, D. Rowbotham13, R.P. Arasaradnam14, C. Marano4
1Icahn School of Medicine at Mount Sinai, Division Of Gastroenterology, New York, United States
2University of California San Diego, San Diego, United States
3University of Calgary, Medicine, Calgary, Canada
4Janssen Research & Development, LLC, Spring House, United States
5Nancy University Hospital, Vandoeuvre, France
6University of Leuven Division of Gastroenterology University Hospitals Leuven, Leuven, Belgium
7Humanitas University/ Humanitas Research Hospital, Milano, Italy
8Cedars-Sinai Medical Center, Los Angeles, United States
9University of Miami, Miller School of Medicine, Crohn’s and Colitis Center, Miami, United States
10Kyorin University School of Medicine, Tokyo, Japan
11Weill Cornell Medical Center, New York Presbyterian, New York, United States
12Concord Repatriation General Hospital, Concord, Australia
13Auckland City Hospital, Gastroenterology & Hepatology, Auckland, New Zealand
14University Hospital Coventry and Warwickshire, Dept. of Gastroenterology, Coventry, United Kingdom
Contact E-Mail Address:bruce.sands@mssm.edu
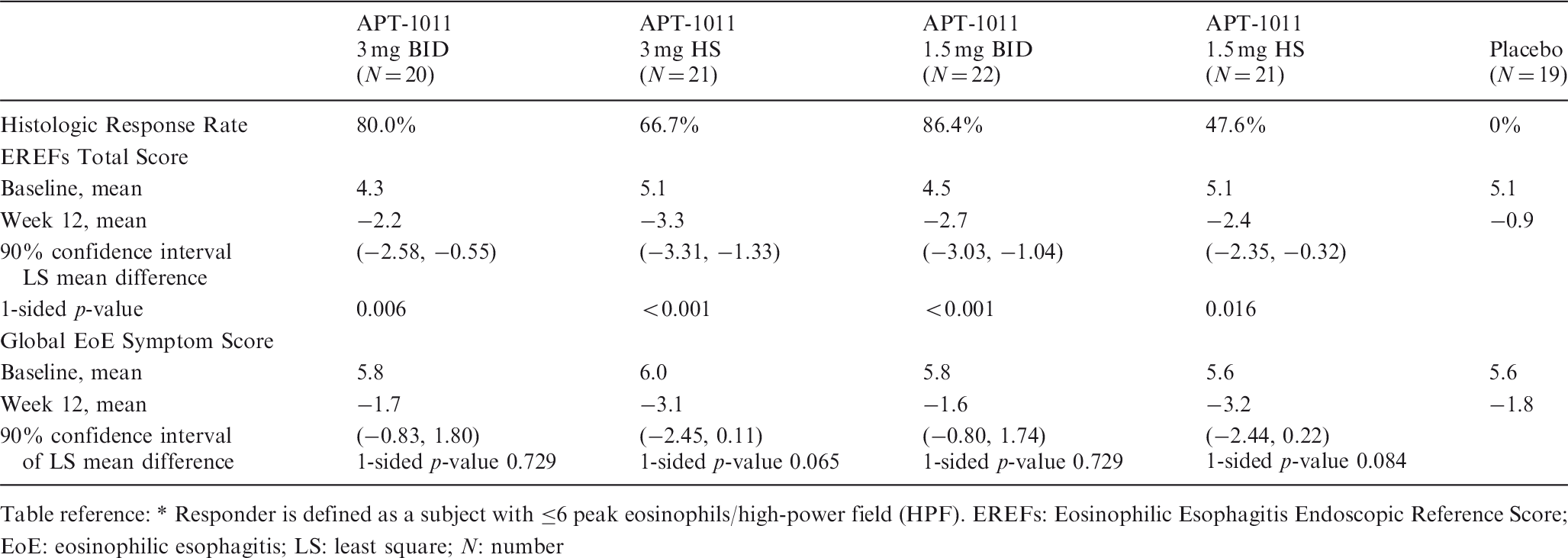
Introduction: Ustekinumab (UST) is a fully human immunoglobulin G1κ mAB antagonist to IL-12/23p40 for moderately to severely active ulcerative colitis (UC). The ongoing UNIFI long-term extension (LTE) evaluates subcutaneous (SC) UST through 220wks of maintenance treatment, with efficacy (wk92) and safety (wk96) results for patients treated in the LTE reported here.
Aims & Methods: 783 patients entered the maintenance study, including 523 intravenous (IV) UST induction responders in the primary population (randomized to SC placebo [PBO]; n = 175, UST 90 mg every 12 weeks [q12w]; n = 172, or UST 90 mg q8w; n = 176). Non-randomized patients included UST delayed responders (patients in clinical response to UST following an IV & SC UST dose) who received SC UST 90 mg q8w and PBO responders who received SC PBO. All patients completing wk44 were eligible to enter the LTE. PBO patients were discontinued after wk44 unblinding. Efficacy measures (i.e. partial Mayo scores and fecal inflammatory biomarkers) were collected every 12wks, thereafter at each dosing visit. Safety was evaluated throughout.
Results: 588 patients were treated in the LTE, including 284 receiving SC UST and 115 receiving SC PBO in the randomized population and 116 UST delayed responders and 73 PBO responders. Rates of discontinuation before wk96 among randomized patients treated in LTE were 8.5% for UST and 40.9% for PBO. Among randomized patients who continued to receive UST in the LTE, percentages of patients in symptomatic remission from wks44 to 92 or up to the time of dose adjustment (as observed) ranged from 72.1% to 81.9% in the q12w group and 77.3% to 87.6% in the q8w group; percentages in partial Mayo remission ranged from 72.1% to 83.8% and 78.0% to 89.4%, respectively. In the ITT analysis of patients who were treated in the LTE, 60.6% and 62.0% of patients in the combined UST group attained symptomatic and partial Mayo steroid-free remission, respectively, at wk92 (Table). Among patients who had achieved clinical remission at maintenance baseline, 73.1% and 74.6% of patients in the combined UST group attained symptomatic and partial Mayo remission, respectively, at both wks 44 and 92.
Among all patients treated in the LTE, those treated with UST had 428.3 patient-years of follow-up vs. 134.0 patient-years for PBO from wks44–92. Safety events per hundred patient-years of follow-up from wks44–92 for combined UST vs. PBO were AEs: 255.68 vs. 267.93, SAEs: 9.34 vs. 12.69, and serious infections: 2.33 vs. 2.99. One UST-treated patient experienced failure to thrive and ultimately expired due to cardiac arrest. Malignancy rates were low and similar between groups: lentigo malignant melanoma in situ (PBO only); BCC (1 PBO with prior UST during induction, 1 q12w and 2 q8w [1 q8w presented with SCC]).
Conclusion: The efficacy of UST in patients with UC was sustained through 92wks. No new safety signals were observed.
[Number of patients with clinical outcomes through Week 92 (ITT analysis); randomized patients in the maintenance study who were treated in the LTE]
Disclosure: This study was funded by Janssen Research & Development, LLC. Drs. Sands, Sandborn, Panaccione, Peyrin-Biroulet, Van Assche, Danese, Targan, Abreu, Hisamatsu, Scherl, Leong, Rowbotham, and Arasaradnam have been investigators for trials sponsored by Janssen and/or have received consulting fees from Janssen. Drs. O’Brien, Zhang, Johanns, Zhou, Tikhonov, and Marano are Janssen employees and own stock and/or stock options in Johnson & Johnson.
Clinical outcome
UST 90 mg SC q12wa (N = 141)
UST 90 mg SC q8wa (N = 143)
UST Combined (N = 284)
Corticosteroid-free symptomatic remission at Week 92b,c,d
83 (58.9%)
89 (62.2%)
172 (60.6%)
Corticosteroid-free partial Mayo remission at Week 92c,d,e
86 (61.0%)
90 (62.9%)
176 (62.0%)
Patients receiving concomitant corticosteroids at maintenance baseline
68 (48.2%)
71 (49.7%)
139 (48.9%)
Corticosteroid-free symptomatic remission at Week 92b,c,d,f
34 (50.0%)
41 (57.7%)
75 (54.0%)
Corticosteroid-free partial Mayo remission at Week 92c,d,e,f
36 (52.9%)
42 (59.2%)
78 (56.1%)
Patients who had achieved clinical remissiong at maintenance baseline
35 (24.8%)
32 (22.4%)
67 (23.6%)
Symptomatic remission at both Week 44 and Week 92b,c,h
27 (77.1%)
22 (68.8%)
49 (73.1%)
Partial Mayo remission at both Week 44 and Week 92c,e,h
27 (77.1%)
23 (71.9%)
50 (74.6%)
aRandomized group at maintenance Week 0, regardless of whether patients had a dose adjustment during the LTE.
bSymptomatic remission is defined as a stool frequency subscore of 0 or 1 and a rectal bleeding subscore of 0. Patients who had both stool frequency and rectal bleeding subscores missing at a visit were considered not to be in symptomatic remission for that visit.
cPatients who had an ostomy or colectomy, or discontinued study agent due to lack of therapeutic effect or due to an AE of worsening of UC or were dose adjusted (only occurred from Week 56 onward) prior to the designated visit, were considered not to be in symptomatic or partial Mayo remission from the time of the event onward.
dPatients who had a missing value in corticosteroid use had their last value carried forward.
ePartial Mayo remission is defined as a partial Mayo score ≤ 2. Patients who had all 3 partial Mayo subscores missing at a visit were considered not to be in partial Mayo remission for that visit.
fDenominator is the number of patients who were receiving concomitant corticosteroids at maintenance baseline.
gClinical remission is defined as a Mayo score ≤2 points, with no individual subscore >1.
hDenominator is the number of patients who had achieved clinical remission at maintenance baseline.
Key: AE, adverse event; SC, subcutaneous; q8w, every 8 weeks; q12w, every 12 weeks; UC, ulcerative colitis.
LB02 NONINFERIORITY OF NOVEL SUBCUTANEOUS INFLIXIMAB (CT-P13) TO INTRAVENOUS INFLIXIMAB (CT-P13) IN PATIENTS WITH ACTIVE CROHN’S DISEASE AND ULCERATIVE COLITIS: WEEK 30 RESULTS FROM A MULTICENTRE, RANDOMISED CONTROLLED PIVOTAL TRIAL
S. Schreiber1, J. Leszczyszyn2, R. Dudkowiak2, A. Lahat3, B. Gawdis-Wojnarska4, A. Pukitis5, M. Horynski6, K. Farkas7, J. Kierkus8, M. Kowalski9, S. Ben-Horin10, B.D. Ye11, S.J. Lee12, S.H. Kim12, M.R. Kim12, H.N. Kim12, W. Reinisch13
3Chaim Sheba Medical Center and Sackler School of Medicine Tel Aviv University, Ramat Gan, Israel
4Twoja Przychodnia – Szczecińskie Centrum Medyczne, Szczecin, Poland
5Pauls Stradins Clinical University Hospital, Riga, Latvia
6Endoskopia Sp. z o.o., Sopot, Poland
7Szent Imre Egyetemi Oktatókórház, Budapest, Hungary
8Centrum Zdrowia Dziecka, Warsaw, Poland
9Centrum Diagnostyczno – Lecznicze Barska Sp. z o.o., Wloclawek, Poland
10University of Tel Aviv Sheba Medical Center, Tel-Hashomer, Israel
11University of Ulsan College of Medicine, Asan Medical Center, Seoul, Korea (Republic of)
12CELLTRION, INC, Incheon, Korea (Republic of)
13Medical University of Vienna, Vienna, Austria
Contact E-Mail Address:s.schreiber@mucosa.de
Introduction: CT-P13 subcutaneous (SC) formulation was developed to augment the flexibility in therapeutic use of infliximab dependent upon need of drug exposure, and also provide patients with expanded, convenient options for chronic therapy which would boost up their quality of life. CT-P13 SC showed comparable efficacy and safety with CT-P13 intravenous (IV) in preliminary studies of Crohn’s disease (CD)1 and rheumatoid arthritis (RA)2. Noninferiority (NI) of CT-P13 SC was demonstrated for efficacy in RA patients3.
Aims & Methods: This randomised, controlled, open-label, multicentre study aimed to demonstrate NI of CT-P13 SC compared with CT-P13 IV in terms of pharmacokinetics (PK), and evaluate efficacy and safety in a mixed population of active CD (Crohn’s Disease Activity Index [CDAI] score of 220 to 450) and ulcerative colitis (UC) (total Mayo score of 6 to 12 with endoscopic subscore of ≥2). After loading doses of IV 5 mg/kg at Weeks 0 and 2, patients were randomised at Week 6 to receive either SC 120 mg (<80 kg) or 240 mg (≥80 kg) every 2 weeks (SC arm), or IV 5 mg/kg every 8 weeks (IV arm). The primary PK endpoint, Ctrough,week22 (pre-dose serum concentration at Week 22), was analysed by using analysis of covariance (ANCOVA). The NI of SC (120 mg and 240 mg combined) was predefined to be met if the lower bound of the 2-sided 90% confidence interval (CI) for the ratio of the geometric least square (LS) means was higher than 80%. Comparative clinical efficacy and safety profiles were assessed for both arms.
Results: In total, 136 patients were enrolled and 131 were randomised (66 to SC and 65 to IV). The primary endpoint, NI of SC compared with IV in terms of Ctrough,week22, was achieved as the lower bound of 90% CI for the ratio of the geometric LS means (786.37–1694.00%) was greater than 80%, with higher geometric LS mean of Ctrough,week22 in SC (20.9844 µg/mL in SC and 1.8181 µg/mL in IV). Frequent administration of small doses and delayed absorption of SC led to more constant exposure compared with IV dosing. Combined clinical remission rates for CD and UC patients at Week 30 were comparable between the 2 arms (66.7% [44/66 patients] in SC and 54.7% [35/64 patients] in IV, p = 0.1620). Mucosal healing at Week 22 was achieved as SES-CD score ≤2 for CD and Mayo endoscopic subscore ≤1 for UC, also comparably between the 2 arms (48.1% [26/54 patients] in SC and 41.0% [16/39 patients] in IV, p = 0.4958). The safety profiles after randomisation were generally comparable between the two arms. Localised injection site reactions occurred more commonly in SC but all were grade 1 or 2 in intensity. The anti-drug antibody positive rate was slightly lower in SC at Week 30 (37.9% [25/66 patients] in SC and 53.8% [35/65 patients] in IV).
Conclusion: Administration of CT-P13 SC resulted in a noninferior drug exposure in comparison with CT-P13 IV in moderately to severely active inflammatory bowel disease patients. CT-P13 SC resulted in adequate trough level, achieving comparable clinical efficacy between the two formulations. It is anticipated that this novel formulation would facilitate and broaden access to efficacious and convenient therapy for the patients.
Disclosure: Schreiber S. receives personal fees from Abbvie, Arena, BMS, Biogen, Celltrion Inc., Celgene, IMAB, Gilead, MSD, Mylan, Pfizer, Fresenius, Janssen, Takeda, Theravance, Provention Bio, Protagonist and Falk, outside the submitted work. Leszczyszyn J., Dudkowiak R., Lahat A., Gawdis-Wojnarska B., Pukitis A., Horynski M., Farkas K., Kierkus J., and Kowalski M. have nothing to disclose. Ben-Horin S. receives consultancy/advisory board fees from Janssen, Takeda, Celltrion Inc., Abbvie, Ferring, Pfizer, GSK and research support from Takeda, Abbvie, Celltrion, Pfizer and Janssen. Ye B. D. receives research grant from Celltrion Inc., consulting fees from Abbvie Korea, Chong Kun Dang Pharm., Daewoong Pharma, Ferring Korea, Janssen Korea, Kangstem Biotech, Kuhnil Pharm., Shire Korea, Takeda Korea, IQVIA, Cornerstones Health, Robarts Clinical Trials Inc. and Takeda, speaking fees from Abbvie Korea, Celltrion Inc., Janssen Korea, Shire Korea, Takeda Korea and IQVIA. Lee S. J., Kim S. H., Kim M. R., and Kim H. N. are employees of Celltrion Inc. Reinisch W. receives personal fees as a speaker for Abbott Laboratories, Abbvie, Aesca, Aptalis, Astellas, Centocor, Celltrion, Danone Austria, Elan, Falk Pharma GmbH, Ferring, Immundiagnostik, Mitsubishi Tanabe Pharma Corporation, MSD, Otsuka, PDL, Pharmacosmos, PLS Education, Schering-Plough, Shire, Takeda, Therakos, Vifor, Yakult, as a consultant for Abbott Laboratories, Abbvie, Aesca, Amgen, AM Pharma, AOP Orphan, Arena Pharmaceuticals, Astellas, Astra Zeneca, Avaxia, Roland Berger GmBH, Bioclinica, Biogen IDEC, Boehringer-Ingelheim, Bristol-Myers Squibb, Cellerix, Chemocentryx, Celgene, Centocor, Celltrion, Covance, Danone Austria, Elan, Eli Lilly, Ernest & Young, Falk Pharma GmbH, Ferring, Galapagos, Genentech, Gilead, Grünenthal, ICON, Index Pharma, Inova, Janssen, Johnson & Johnson, Kyowa Hakko Kirin Pharma, Lipid Therapeutics, LivaNova, Mallinckrodt, Medahead, MedImmune, Millenium, Mitsubishi Tanabe Pharma Corporation, MSD, Nash Pharmaceuticals, Nestle, Nippon Kayaku, Novartis, Ocera, Otsuka, Parexel, PDL, Periconsulting, Pharmacosmos, Philip Morris Institute, Pfizer, Procter & Gamble, Prometheus, Protagonist, Provention, Robarts Clinical Trial, Sandoz, Schering-Plough, Second Genome, Seres Therapeutics, Setpointmedical, Sigmoid, Takeda, Therakos, Tigenix, UCB, Vifor, Zealand, Zyngenia, and 4SC, as an advisory board member for Abbott Laboratories, Abbvie, Aesca, Amgen, AM Pharma, Astellas, Astra Zeneca, Avaxia, Biogen IDEC, Boehringer-Ingelheim, Bristol-Myers Squibb, Cellerix, Chemocentryx, Celgene, Centocor, Celltrion, Danone Austria, Elan, Ferring, Galapagos, Genentech, Grünenthal, Inova, Janssen, Johnson & Johnson, Kyowa Hakko Kirin Pharma, Lipid Therapeutics, MedImmune, Millenium, Mitsubishi Tanabe Pharma Corporation, MSD, Nestle, Novartis, Ocera, Otsuka, PDL, Pharmacosmos, Pfizer, Procter & Gamble, Prometheus, Sandoz, Schering-Plough, Second Genome, Setpointmedical, Takeda, Therakos, Tigenix, UCB, Zealand, Zyngenia, and 4SC, and has received research funding from Abbott Laboratories, Abbvie, Aesca, Centocor, Falk Pharma GmbH, Immundiagnsotik, MSD.
References
Ye BD, et al. Tu1715 A Novel Formulation of CT-P13 (Infliximab Biosimilar) for Subcutaneous Administration: 1-Year Result from a Phase I Open-Label Randomized Controlled Trial in Patients with Active Crohn's Disease. Gastroenterology 2019; 156(6).
Yoo DH, et al. Fri0128 A Novel Formulation of CT-P13 (Infliximab Biosimilar) for Subcutaneous Administration: 1-Year Results from a Part 1 of Phase I/III Randomized Controlled Trial in Patients with Active Rheumatoid Arthritis. Poster Presentations. 2019.
Westhovens R, et al. Sat0170 A Novel Formulation of CT-P13 for Subcutaneous Administration: 30 Week Results from a Part 2 of Phase I/III Randomized Controlled Trial in Patients with Rheumatoid Arthritis. 2019.
Note: Randomisation at Week 6 to treatment assignment was stratified by concomitant use of immunomodulators, disease (CD or UC), clinical response at Week 6 (responder or nonresponder by CDAI-70 for CD and partial Mayo score for UC), and body weight at Week 6 (<80 kg or ≥80 kg).
1. Patients with decrease in CDAI score of 70 points or more from the baseline value.
2. Patients with CDAI score of less than 150 points.
3. Partial Mayo score was composed of stool frequency, rectal bleeding and physician’s global assessment.
4. Patients with decrease in partial Mayo score from baseline at least 2 points, with an accompanying decrease in the subscore for rectal bleeding of at least 1 point, or an absolute subscore for rectal bleeding of 0 or 1.
5. Patients with partial Mayo score of 1 point or lower.
[Clinical results up to Week 30]
LB03 MICROVILLI LENGTH PREDICTS CLINICAL RESPONSE TO USTEKINUMAB IN CROHN’S PATIENTS FROM THE UNITI-2 TRIAL
K. Van Dussen1, K. Li2, K. Simpson3, B. Claggett4, J. Friedman5, J. Perrigoue5, T. Stappenbeck6
1University of Cincinnati College of Medicine, Pediatrics, Cincinnati, United States
2Janssen Research & Development, Spring House, United States
3Washington University School of Medicine, Pathology and Immunology, St. Louis, United States
4Harvard Medical School, Boston, United States
5Janssen Research & Development, Immunology, Spring House, United States
6Washington University Medical School, Department of Pathology and Immunology, St. Louis, United States
Contact E-Mail Address:stappenb@wustl.edu
Introduction: Mucosal biomarkers have been shown to identify responders to anti-TNF and anti-integrin therapies in Crohn’s Disease (CD) patients. We have recently identified a novel epithelial biomarker of malabsorption, microvilli (MV) length, to be significantly reduced in CD patients. We hypothesize that malabsorption contributes to the development of intestinal inflammation and therefore may have predictive value for clinical and endoscopic response to biologic therapy in CD patients. Ustekinumab (UST) is an anti-IL12/23 monoclonal antibody approved for the treatment of CD.
Aims & Methods: The aim of this study was to determine the predictive value of pre-treatment ileal biopsy MV length for clinical and endoscopic response to UST in CD patients. Biopsy samples from the American cohort of UNITI-2 trial patients were stained for H&E and analyzed for MV length. Clinical response, defined as a reduction of CDAI of ≥100 points from pre-treatment baseline, clinical remission, defined as CDAI < 150, and endoscopic response, defined as ≥50% reduction from baseline SES-CD score, at 8 weeks post-induction were assessed. The primary outcome was clinical response to UST stratified by reduction in pre-treatment MV length, using a threshold of 1.7 µm (normal above, low below). Outcomes were compared using Fisher exact tests for subgroups and likelihood ratio tests for tests of effect modification.
Results: A total of 106 CD patients from UNITI-2 (placebo = 36, UST = 70) with biopsies were stained, of which 95 patients (90%) had sufficient samples for analysis of MV length. There were no significant differences in baseline patient characteristics between placebo- (N = 36) and UST-treated (N = 70) patients (Table 1). As a continuous variable, MV length was an effect modifier of clinical response to UST therapy (p = 0.043).The overall clinical response was significantly higher in the UST-treated group compared with placebo: 65% (40/62) vs. 39% (13/33, p = 0.03). The greatest therapeutic effect and differences were seen in the normal MV length group: clinical response UST vs. placebo, 85% (11/13) vs. 20% (1/5, p = 0.02); corresponding rates in the low MV length group were 59% (28/49) vs. 46% (12/28, p = 0.17). Similar results were seen with respect to endoscopic response: normal MV length group (75% [6/8] vs. 20% [1/5]), compared with low MV length group (65% [24/35] vs. 48% [11/23]).
Conclusion: Pre-treatment ileal biopsy MV length was predictive of clinical and endoscopic response to UST in a cohort of UNITI-2 trial patients, with the greatest therapeutic effects observed in patients with normal MV length.
Disclosure: Nothing to disclose.
Baseline Patient Characteristics
Placebo (N = 36)
Ustekinumab (N = 70)
p-value
Mean age, years ± SD
43 ± 14
38 ± 12
0.10
Female gender, N (%)
18 (50)
40 (57)
0.48
Ethnicity, N (%) Caucasians
32 (89)
60 (86)
0.42
African Americans
1 (3)
6 (9)
Asians or others
3 (9)
4(5)
Average BMI ± SD, kg/m2
26.2 ± 6.0
26.0 ± 5.8
0.46
Disease duration (Years)
6.4 [2.4, 15.9]
4.0 [1.5, 13.4]
0.15
Disease phenotype, N (%) Fistulizing
2 (6)
5 (7)
0.76
Fibrostenotic
8 (22)
14 (20)
0.18
Disease location, N (%) Ileum only
12 (33)
17 (24)
0.42
Colon only
19 (53)
34 (49)
Ileum and colon
5 (14)
18(26)
Prior anti-TNF exposure
14 (48 %)
27 (40%)
0.43
C-reactive protein (CRP)
2.8 [1.0, 14.4]
5.1 [2.4, 10.6]
0.55
LB04 COMBINED ILEAL BIOMARKERS CAN DISCRIMINATE RESPONDERS FROM NON-RESPONDERS TO VEDOLIZUMAB THERAPY IN CROHN’S DISEASE
M.T. Osterman1, E.M. Davis2, I.O. Gordon3, K. Simpson4, M. Ciorba5, S.C. Glover6, B.P. Abraham7, E. Yee8, F. Allard8, B. Claggett9, B. Shen10, T.S. Stappenbeck4, J. Liu2
1University of Pennsylvania, Medicine, Philadelphia, United States
2University of Arkansas for Medical Sciences, Medicine, Little Rock, United States
3Cleveland Clinic Foundation, Cleveland, Pathology, Cleveland, United States
4Washington University School of Medicine, St Louis, United States
5Washington University School of Medicine, Medicine, St. Louis, United States
6University of Florida, Gainesville, United States
7Baylor College of Medicine, Houston, United States
8University of Arkansas for Medical Sciences, Pathology, Little Rock, United States
9Brigham and Women’s Hospital, Little Rock, United States
10Cleveland Clinic Foundation Digestive Disease Institute, Cleveland, United States
Contact E-Mail Address:jjliu@uams.edu
Introduction: Several mucosal biomarkers have been identified to hold predictive values for biologic therapies in Crohn’s disease (CD). Vedolizumab is an anti-integrin monoclonal antibody approved for the treatment of CD. We have recently shown that ileal epithelial cell pyroptosis, a biomarker of innate immune activation, can help identify responders to vedolizumab therapy in CD. Ileal epithelial cell microvilli length (MVL), a novel biomarker of malabsorption, was shown to be significantly reduced in CD patients and may also have predictive value for biologic treatment. The utility of using the combination of these two biomarkers to predict response to vedolizumab therapy in CD is unknown.
Aims & Methods: The aim of this study was to determine the relationship between ileal epithelial cell pyroptosis and ileal MVL and whether the combination of these two biomarkers may improve the identification of responders and non-responders to vedolizumab in CD. Patients aged 18 to 80 years with known diagnosis of CD from five American IBD centers with pre-vedolizumab ileal biopsies were enrolled. Clinical response, defined as a reduction of Harvey-Bradshaw Index (HBI) of ≥5 points from pre-treatment baseline, and clinical remission, defined as HBI <5, was determined ≥6 months after therapy. Biopsy samples were sectioned and stained for pyroptosis using the Maximus Biological Assay kit (Maximus Diagnostics LLC). Ileal MVL was taken as the average of 50 measurements obtained from 10 intact villi per patient on H&E stained sections, at 100× magnification by a blinded pathologist. Clinical response rates to vedolizumab stratified by pre-treatment MVL were examined. The relationship between ileal pyroptosis and MVL was described using Spearman’s correlation. The value of ileal pyroptosis and/or MVL thresholds alone or in combination for prediction of clinical response was compared using Fisher’s exact test.
Results: 55 CD patients with pre-treatment ileal biopsies were enrolled; 43 had adequate samples for analysis of both epithelial cell pyroptosis and MVL. The overall clinical response rate was 58% (25/43). There were no significant differences in baseline patient characteristics, disease characteristics, and concomitant medication use between responders (N = 25) and non-responders (N = 18). As a single biomarker, ileal MVL range of 1.35–1.55 µm was associated with a response rate of 82% (14/17), versus 44% (7/16) for <1.35 µm, and 40% (4/6) for >1.55 µm (p = 0.038). There was no significant correlation between ileal MVL with ileal epithelial cell pyroptosis (Spearman’s rho = +0.11, p = 0.50). The combination criteria of ileal pyroptosis level < 14 positive cells/1000 IECs or MVL range of 1.35–1.55 µm could discern responders with response rate of 78% (21 of 27 positive for criteria) from non-responders with response rate of 25% (4 of 16 negative for criteria, p = 0.001), comparable to placebo response observed in clinical trials.
Conclusion: Ileal MVL and epithelial cell pyroptosis are two independent mucosal biomarkers of clinical response to vedolizumab in CD patients. The combination of two biomarkers can discriminate responders from non-responders to vedolizumab in Crohn’s disease patients.
Disclosure: Elisabeth Davis and Julia Liu are patent holders of Maximus Diagnostics LLC.
LB05 REAL-WORLD EXPERIENCE WITH USTEKINUMAB IN PAEDIATRIC CROHN’S DISEASE. A MULTICENTRE RETROSPECTIVE STUDY FROM PAEDIATRIC IBD PORTO GROUP OF ESPGHAN
G. Pujol Muncunill1, V.M. Navas-López2, O. Ledder3, S. Cohen4, M. Lekar3, D. Turner5, K.-L. Kolho6, A. Levine7, N. Croft8, J. Bronsky9, D.S. Souval10, A. Assa11, R. Harris12, F. Kiparissi13, M. Aloi14, N. Afzal15, C. TZIVINIKOS16, J. Barrios17, C. Norden18, M.J. Balboa Vega19, S. Buderus20, A. Fernández de Valderrama21, L. de Ridder22, R. Garcia-Romero23, E. Medina24, C. Sánchez25, M. Velasco26, S. Vicente27, D.C. Wilson28, S. Naik29, O. Hradsky30, L. Cococcioni31, F.J. Martin De Carpi32; ESPGHAN IBD Porto Group
1Hospital Sant Joan de Déu, Unit for the Comprehensive Care of Paediatric Inflammatory Bowel Disease. Paediatric Gastroenterology, Hepatology and Nutrition Department, Esplugues del Llobregat, Spain
2Hospital Regional Universitario de Malaga, Malaga, Spain
3Shaare Zedek Medical Center, Jerusalem, Israel
4Tel Aviv MC, Tel Aviv, Israel
5Shaare Zedek Medical Center, Pediatric GI, Jerusalem, Israel
6University of Helsinki and Helsinki University Hospital and Tampere University and Tampere Universit, Faculty of Medicine and Biosciences, Tampere, Finland
7Wolfson Medical Center, Paediatric Gastroenterology, Holon, Israel
8The Royal London Children’s Hospital, London, United Kingdom
9Faculty Hospital Motol Dept. of Pediatrics, Prague 5, Czech Republic
10Sheba Medical Center, Tel Hashomer, Israel
11Schnider Children’s Medical Center, Dr., Petach Tikva, Israel
12Royal Hospital for Children, Glasgow, United Kingdom
13Great Ormond Street Hospital NHS Foundation Trust, Gastroenterology, London, United Kingdom
14Sapienza University of Rome Dept. of Pediatric Gastroenterology SIGENP IBD Group, Pediatric Gastroenterology And Liver Unit, Rome, Italy
15Southampton Children’s Hospital, Southampton, United Kingdom
16Al Jalila Children’s Specialty Hospital, Paediatric Gastroenterology, DUBAI, United Arab Emirates
17Hospital Universitario de Fuenlabrada, Madrid, Spain
18Hvidovre Hospital, Copenhagen, Denmark
19Hospital Virgen de la Macarena, Sevilla, Spain
20GFO-Kliniken Bonn, St. Marien-Hospital, Pediatrics, Bonn, Germany
21Hospital Universitario de Burgos, Burgos, Spain
22Erasmus MC Rotterdam Sophia Children’s Hospital Dept. of Pediatric Gastroenterology, Rotterdam, Netherlands
23Hospital Universitario Miguel Servet, Zaragoza, Spain
24Hospital 12 de Octubre, Pediatrics, Madrid, Spain
31Great Ormond Street Hospital, London, United Kingdom
32Hosp. Sant Joan De Deu, Unit for the Comprehensive Care of Paediatric Inflammatory Bowel Disease. Paediatric Gastroenterology, Hepatology and Nutrition Department, Barcelona, Spain
Contact E-Mail Address:pumun@hotmail.com
Introduction: Ustekinumab is a fully human monoclonal antibody that blocks the p40 subunit of interleukin 12/23. Ustekinumab is effective for induction and maintenance of remission in adult Crohn’s disease (CD) but data in paediatric CD is scarce. This study aims to describe the effectiveness and safety of ustekinumab in refractory paediatric CD patients in a European multi-centre cohort in real-life practice.
Aims & Methods: The aim of our study was to describe the effectiveness and safety of ustekinumab in refractory paediatric CD patients in a European multi-centre cohort in real-life practice.
Retrospective review of children with CD (2–18 years old) who were treated with ustekinumab (at least one dose) from centres worldwide affiliated with the Paediatric IBD Interest and Porto group of ESPGHAN. Primary outcome was corticosteroid (CS) and Exclusive Enteral Nutrition (EEN)-free remission (defined by wPCDAI (weighted Paediatric Crohn’s Disease Activity Index < 12.5) and safety at week 6 and 52.
Results: A total of 101 patients (23 centres, 10 countries) were included (51 males (50.4%), mean age at ustekinumab initiation 15.4 years (IQR 12.7–17.2), mean previous disease duration 4.3 years (IQR 2.5–6.8)). Sixty percent of the patients had ileocolonic involvement (L3) and 24.8% perianal disease. The median wPCDAI at ustekinumab initiation was 38.7 (IQR 25–57.5). Ninety-six percent were previously treated with anti-TNF (60.4% with two of them) and 22% had received vedolizumab. The most used induction strategy was an intravenous dose of 6 mg/kg (IQR 5–6) and in 79% of the patients maintenance strategy was based in 90 mg doses sc every 8 weeks. At week 6 (n = 74) 38% of these patients achieved the primary outcome and at week 52 (n = 49) 50% of patients maintained remission. We did not find any predictive factor associated with ustekinumab-induced clinical remission in our cohort in the multivariate analysis. No malignancies were reported during the follow-up (mean duration of ustekinumab treatment: 14.1 months (IQR 9.1–18.9)). Six minor adverse events (three infections, one infusion reaction, one abnormal laboratory result, one vasculitis of the tongue) and seven patients with clinical deterioration due to the disease (three of them requiring hospitalization) were reported. One patient died by an unrelated ustekinumab cause during the follow-up.
Conclusion: This is the largest cohort of ustekinumab use in paediatric CD patients thus far. Despite its retrospective nature and the lack of standardized treatment, we demonstrate that ustekinumab was effective and safe in a sub-cohort of refractory paediatric CD patients at short and mid-term follow-up. Larger cohorts as well as prospective studies are needed to confirm these results at long-term follow-up.
Disclosure: Nothing to disclose.
LB06 ORAL ABX464 QD IS SAFE AND EFFICACIOUS DURING 52 WEEKS OPEN LABEL MAINTENANCE FOLLOWING A PLACEBO-CONTROLLED INDUCTION STUDY IN ULCERATIVE COLITIS PATIENTS
S. Vermeire1, X. Hebuterne2, P. Napora3, M. Wisniewska Jarosinska4, G. Kiss5, A. Bourreille6, P. Zajac7, J. Nitcheu8, P. Gineste8, H. Ehrlich8, J.-M. Steens9
1University Hospital Leuven – Dept. of Gastroenterology, University Hospital Leuven; Leuven/BE – Dept, Dept. of Gastroenterology, Leuven, Belgium
2Hospital Archet 2, Nice, Department of Gastroenterology and Clinical Nutrition, Nice, France
3Piotr Napora Centrum Badan Klinicznych Lekarze Sp.p, Wraclow, Poland
4Department of Gastroenterology Mediacal University of Lodz, Dr., Lodz, Poland
5Hajdú-Bihar Megyei Önkormányzat Kenézy Gyula Kórház, Debrecen, Hungary
6Hopital Hotel Dieu et HME, Gastroenterologie, Nantes Cedex, France
Introduction: Despite the availability of new drugs in IBD, there is still a high unmet medical need for patients suffering from ulcerative colitis. ABX464 has potent anti-inflammatory properties impacting the expression of miR124. A phase 2 a study was conducted in patients with moderate to severe ulcerative colitis intolerant and/or refractory to existing treatments. Following a randomised double-blind placebo-controlled induction phase, patients were eligible to be rolled over in an open label study with oral ABX464 (50 mg) for a total of 52 weeks
Aims & Methods: In the placebo-controlled induction study 32 patients from 15 European centres were randomized 2:1 to ABX464 50 mg QD orally or placebo for 8 weeks. 22 Patients were enrolled in the open label ABX464 (50 mg) 52 weeks maintenance study. Safety and efficacy endpoints were similar than those used in the induction study. Primary endpoint was safety of ABX464 and key secondary endpoints included clinical remission (rectal bleeding sub-score = 0 and a Mayo endoscopic sub-score ≤ 1 and at least one-point decrease in stool frequency sub score from baseline to achieve a stool frequency sub-score ≤1), endoscopic improvement (Mayo endoscopic score of 0 or 1), and clinical response.
Results: Safety was good and the efficacy results of the induction study are summarised in the table.
22 patients from five different sites were included in the 52 weeks open label ABX464 50 mg maintenance study.
Safety and tolerability remained unchanged. A total of 19 patients (86%) completed 52 weeks of therapy and entered a second maintenance year of ABX464.Three patients dropped out during the study (two because absence of clinical efficacy, one for headache grade 2). In 16/19 patients endoscopy was performed at week 52 ± 3 months.
16/16 (100%) patients had a Mayo endoscopic subscore of 0 or 1.
When combined with rectal bleeding and stool frequency assessed at 52 weeks, this resulted in 12/16 (75 %) patients in clinical remission
• From the seven patients who were in clinical remission at the end of the induction study, five remained in clinical remission and two had no endoscopy performed but still showed clinical response
• From the 12 patients who were not in clinical remission at the end of the induction phase, seven patients achieved clinical remission at week 52, one had no endoscopy performed but still had clinical benefit and the four patients who did not achieve clinical remission at week 52 still showed clinical benefit
• Total and Partial Mayo score were respectively reduced from 4.50 and 2.68 at the start of maintenance to 1.94 and 1.47 at 52 weeks
• Median faecal calprotectin levels decreased from 1044.4 µg/g at baseline of the induction to 153 µg/g at week 8 (=baseline of the maintenance study) to 27.9 µg/g at week 52.
• Treatment with ABX464 resulted in >200 fold expression of miR124 levels
Conclusion: The open-label ABX464 maintenance study showed persistent good safety and tolerability of 50 mg given ORALLY during 52 weeks, and established the potential of this drug to further reduce TMS and PMS, normalising faecal calprotectin and keeping miR124 over expressed resulting in 75% of patients achieving clinical remission and to keep clinical benefit in all patients.
Disclosure: Receipt of grants/research supports: MSD, Abbvie, Takeda, Janssen, Pfizer
Receipt of honoraria or consultation fees: AbbVie, MSD, Takeda, Ferring, Genentech/Roche, Shire, Pfizer Inc, Galapagos, Mundipharma, Hospira, Celgene, Second Genome, Progenity, Lilly, Arena, Gilead and Janssen.
Participation in a company sponsored speaker's bureau: AbbVie, MSD, Takeda, Ferring, Hospira, Pfizer, Janssen, and Tillots.
Efficacy after 8 weeks induction
ABX464 (n = 20)
Placebo (n = 9)
p-value
Clinical Remission
35%
11%
0.16
Endoscopic Improvement
50%
11%
0.03
Clinical Response
70%
33%
0.06
Total Mayo Score Reduction
–53%
–27%
0.03
Partial Mayo Score Reduction
–62%
–32%
0.02
Faecal Calprotectin decrease >50%
75%
50%
miRNA124 fold expression
7.69
1.46
0.004
LB07 EFFICACY OF USTEKINUMAB SUBCUTANEOUS MAINTENANCE TREATMENT BY INDUCTION-DOSE SUBGROUP IN THE UNIFI STUDY OF PATIENTS WITH ULCERATIVE COLITIS
S. Danese1, B.E. Sands2, W.J. Sandborn3, C. Marano4, C. O'Brien4, H. Zhang4, J. Johanns4, L. Peyrin-Biroulet5, E. Scherl6, T. Hisamatsu7, R. Panaccione8
1Humanitas University/Humanitas Research Hospital, Milano, Italy
2Icahn School of Medicine at Mount Sinai, Division Of Gastroenterology, New York, United States
3University of California San Diego, San Diego, United States
4Janssen Research & Development, LLC, Spring House, United States
5Nancy University Hospital, Nancy, France
6Weill Cornell Medicine, Jill Roberts Center for Inflammatory Bowel Disease, New York, United States
7Kyorin University, Internal Medicine, Tokyo, Japan
8University of Calgary, Medicine, Calgary, Canada
Contact E-Mail Address:silvio.danese@hunimed.eu
Introduction: Ustekinumab (UST) induction and maintenance was safe and effective in patients with moderately to severely active ulcerative colitis (UC) in the UNIFI study.1,2 Previous presentation of the maintenance study results reported data for all patients who were randomized to subcutaneous (SC) maintenance treatment regardless of induction treatment received. Here, we present results from the maintenance study by subgroups of individual UST IV induction dose.
Primary and key secondary endpoints at Week 44 in patients who received UST at induction baseline and were in clinical response at Week 8
UST 130 mg IV induction
UST ∼6 mg/kg IV induction
PBO SC maintenancea
UST SC maintenanceb
PBO SC maintenancea
UST SC maintenanceb
Patients who were in clinical responsec to UST at Week 8
58
114
69
139
Patients in clinical remissiond at Week 44
19 (32.8%)
43 (37.7%) p = 0.496
14 (20.3%)
68 (48.9%) p < 0.001
Patients who maintained clinical responsec through Week 44 e
29 (50.0%)
76 (66.7%) p = 0.034
31 (44.9%)
108 (77.7%) p < 0.001
Patients with endoscopic improvementf at Week 44
20 (34.5%)
53 (46.5%) p = 0.119
19 (27.5%)
77 (55.4%) p < 0.001
Patients in clinical remissiond and not receiving corticosteroids at Week 44
19 (32.8%)
40 (35.1) p = 0.747
13 (18.8%)
68 (48.9%) p < 0.001
Patients in clinical remissiond at maintenance baseline
20 (34.5%)
30 (26.3%)
16 (23.2%)
33 (23.7%)
Patients who maintained clinical remissiond,g,h through Week 44
9 (45.0%)
16 (53.3%) p = 0.773
5 (31.3%)
21 (63.6%) p = 0.065
aPatients who were in clinical response to ustekinumab IV induction dosing and were randomized to placebo SC on entry into this maintenance study.
bPatients who were in clinical response to ustekinumab IV induction dosing and were randomized to ustekinumab SC 90 mg q12w or q8w on entry into this maintenance study.
cClinical response was defined as a decrease from induction baseline in the Mayo score by ≥30% and ≥3 points, with either a decrease from induction baseline in the rectal bleeding subscore ≥1 or a rectal bleeding subscore = 0 or 1.
dClinical remission was defined as a Mayo score ≤2 points, with no individual subscore >1.
ePatients who maintained clinical response did not show a loss of clinical response through Week 44.
fEndoscopic improvement was defined as a Mayo endoscopy subscore = 0 or 1.
gPatients who maintained clinical remission did not show a loss of clinical remission through Week 44.
hDenominator is subjects in clinical remission at maintenance baseline.
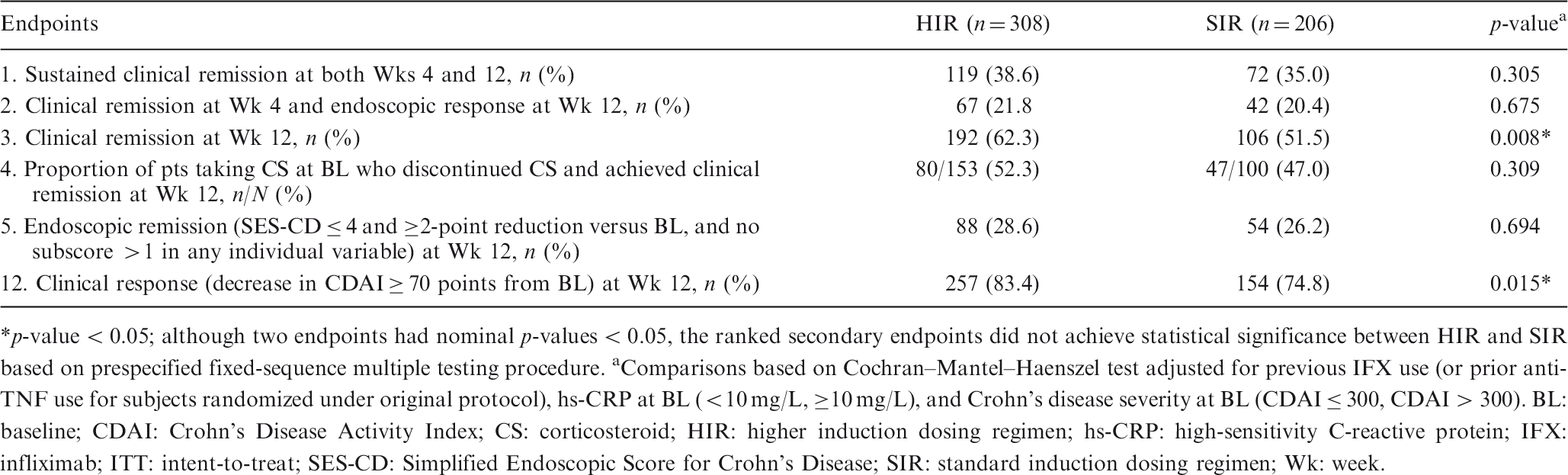
Aims & Methods: UC patients who failed conventional or biologic therapy (including anti-TNFs and/or vedolizumab) were randomized to receive a single IV induction dose of placebo (PBO) or UST 130 mg or ∼6 mg/kg (260 mg for weight ≤55 kg, 390 mg for weight >55 kg and ≤85 kg, or 520 mg for weight >85 kg) at induction baseline (Week 0). Patients who were in clinical response 8 weeks after UST IV induction were randomized to SC PBO, UST 90 mg q12w, or UST 90 mg q8w at maintenance baseline. The primary endpoint in the maintenance study was clinical remission at maintenance Week 44 (52 weeks after IV UST induction); key secondary endpoints were maintenance of clinical response, endoscopic improvement, corticosteroid-free clinical remission, and maintenance of clinical remission among patients who achieved clinical remission at maintenance baseline. Maintenance data are presented for subgroups of patients by induction dose (130 mg or ∼6 mg/kg), comparing maintenance UST SC with PBO. The maintenance study was not powered to compare clinical outcomes in these subgroups.
Results: Among patients who received UST ∼6 mg/kg IV induction at Week 0 and responded at Week 8, greater proportions of patients who were randomized to receive UST SC maintenance were in clinical remission at Week 44, maintained clinical response through Week 44, achieved endoscopic improvement at Week 44, and were in clinical remission and not receiving corticosteroids at Week 44 compared with patients randomized to PBO SC maintenance (Table 1). Among patients who received UST 130 mg IV induction at Week 0 and responded at Week 8, differences between UST and PBO SC maintenance were smaller than those observed in the ∼6 mg/kg group. Among patients who were in clinical remission at maintenance baseline, numeric differences between UST and PBO SC maintenance groups in the percentage of patients who maintained clinical remission through Week 44 were also seen, with larger differences observed in patients who received ∼6 mg/kg induction than those who received the 130 mg induction.
Conclusion: Patients who received UST ∼6 mg/kg IV induction followed by UST SC maintenance had better clinical outcomes after 1 year than those who received UST ∼6 mg/kg IV induction followed by PBO SC maintenance. These results suggest that the ∼6 mg/kg is the optimal induction dose, when combined with UST maintenance treatment, to achieve clinical benefit in patients with moderately to severely active UC.
Disclosure: This study was funded by Janssen Research & Development, LLC. Drs. Danese, Sands, Sandborn, Peyrin-Biroulet, Scherl, Hisamatsu, and Panaccione have been investigators for trials sponsored by Janssen and/or have received consulting fees from Janssen. Drs. Marano, O’Brien, Zhang, and Johanns are Janssen employees and own stock and/or stock options in Johnson & Johnson.
References
Sand BE, Sandborn WJ, Panaccione R, et al. Safety and efficacy of ustekinumab induction therapy in patients with moderate to severe ulcerative colitis: Results from the Phase 3 UNIFI Study. Presented at ACG 2018, October 9, 2018, Philadelphia, PA.SandbornWJSandsBEPanaccioneR, et al.OP37 Efficacy and safety of ustekinumab as maintenance therapy in ulcerative colitis: Week 44 results from UNIFI. Journal of Crohn’s and Colitis2019; 13(Suppl 1): S25–S26.
16:00–17:30 / C2
Hot news from endoscopy
LB08 PERSONALISED DYNAMIC SURVEILLANCE STRATEGIES IN BARRETT’S OESOPHAGUS: A MULTICENTRE PROSPECTIVE COHORT STUDY
C.A.M. Roumans1,2,3, A. Tomer3,4, I. Lansdorp-Vogelaar2, K. Biermann5, M.J. Bruno1, E.W. Steyerberg2,6, M.C.W. Spaander1, D. Rizopoulos4; on behalf of the ProBar study group7
1Erasmus MC, University Medical Center Rotterdam, Department of Gastroenterology & Hepatology, Rotterdam, Netherlands
2Erasmus MC, University Medical Center Rotterdam, Department of Public Health, Rotterdam, Netherlands
4Erasmus MC, University Medical Center Rotterdam, Department of Biostatistics, Rotterdam, Netherlands
5Erasmus MC, University Medical Center Rotterdam, Department of Pathology, Rotterdam, Netherlands
6Leiden University Medical Center, Department of Biomedical Data Sciences, Leiden, Netherlands
7Erasmus MC, University Medical Center Rotterdam, Rotterdam, Netherlands
Contact E-Mail Address:c.roumans@erasmusmc.nl
Introduction: In Barrett’s oesophagus (BO), guidelines recommend fixed surveillance intervals based on the histological diagnosis of the most recent endoscopy. If low-grade dysplasia (LGD) is detected, surveillance is intensified. Current surveillance strategies lead to many unnecessary endoscopies for patients who do not develop neoplastic progression. The longitudinal course (i.e. successive measurements) of histological diagnosis and immunohistochemical biomarkers has been shown to be associated with neoplastic progression. This enables a more personalised and risk-based surveillance strategy. In this study we aimed (1) to define the performance of personalised dynamic surveillance intervals in terms of harms, benefits, and costs, and (2) to compare these with fixed intervals, as currently recommended by the guideline.
Aims & Methods: We fitted a multivariate joint model with LGD, p53, and SOX2 as predictors for high-grade dysplasia (HGD) or oesophageal adenocarcinoma (OAC), analysing a multicentre prospective cohort study of 631 BO patients.1 The discriminative power of the model between patients with low and high neoplastic progression risk was estimated by using the area under the receiver operating curve (AUC), adjusted for optimism. This prediction model was used to define risk-based surveillance schedules with four neoplastic progression risk cut-offs: 1.5%, 2.0%, 2.5%, and 3.0%. The next endoscopy could be scheduled at the time point that the neoplastic progression risk reached this threshold. The delay in years between true neoplastic progression time, rather than the time of detection, was estimated by simulation for each cut-off. Besides, we estimated the number of surveillance endoscopies and mean costs per patient. All parameters were compared with the performance of the fixed schedule as currently recommended by the guideline.
Results: The personalised prediction model could discriminate well between low and high-risk patients with AUC ranging 0.80–0.88 during follow-up. The strategy with the best balance between harms (number of endoscopies) and benefits (finding neoplastic progression early) was the personalised schedule, in which the next surveillance endoscopy would be planned at the time point that the neoplastic progression risk was 2.5% (Table). Compared with the current guideline of fixed intervals, in this personalised strategy a median of three endoscopies per patient in a lifetime could be saved in those who would never progress to HGD/OAC. At the same time, there was no need to perform more endoscopies or to have a longer delay in detection of HGD/OAC, in patients who would progress. This indicated a reduction in mean costs of €2,733 per patient in our cohort (€1.7 million in total) compared with current guideline recommendations.
Conclusion: Given the good discriminative power between patients with a low and high neoplastic progression risk, and an excellent balance between harms and benefits, this personalised dynamic surveillance strategy is likely to reduce the patient and healthcare burden in BO surveillance. After further validation, this personalised strategy may be worthwhile to be implemented in the current guideline.
Disclosure: Nothing to disclose.
Performance of fixed and personalised schedule, represented per patient.
Per patient:
Fixed
Risk 1.5%
Risk 2.0%
Risk 2.5%
Risk 3.0%
Median n° endoscopies (IQR)
Non-progressors
7 (7–9)
5 (4–6)
4 (4–5)
4 (4–5)
4 (4–4)
Progressors
6 (4–7)
8 (4–14)
7 (3–13)
6 (3–12)
5 (3–11)
Median delay in years (IQR)
0.9 (0.5–1.8)
0.4 (0.2–0.6)
0.4 (0.2–0.8)
0.4 (0.2–1.0)
0.5 (0.2–1.3)
Mean total costs
€10,174
€8,075
€7,662
€7,441
€7,276
Reference
RoumansC.A.M.SpaanderM.C.W.Lansdorp-VogelaarI, et al.Prediction in surveillance of Barrett’s esophagus: The effect of multiple measurements on the estimated neoplastic progression risk. United European Gastroenterol Journal2018; 6: Supplement (A84)–Supplement (A84).
LB09 FIRST IN-HUMAN (FIH) STUDY ON THE SAFETY, TOLERABILITY AND EFFICACY OF THE AQUAMEDICAL FOCAL RF-VAPOR ABLATION (RFVA) SYSTEM, FOR THE ERADICATION OF BARRETT’S ESOPHAGUS (BE)
S. van Munster1, R. Pouw1, B.L.A.M. Weusten2,3, J. Bergman1
1Amsterdam UMC, Gastroenterology and Hepatology, Amsterdam, Netherlands
2St Antonius Hospital, Department of Gastroenterology and Hepatology, Nieuwegein, Netherlands
3University Medical Center Utrecht, Gastroenterology and Hepatology, Utrecht, Netherlands
Contact E-Mail Address:s.n.vanmunster@amc.uva.nl
Introduction: Radiofrequency ablation (RFA) has become the preferred modality for ablation of dysplastic BE. Although highly effective, RFA has drawbacks such as the need for multiple deployment steps; for contact with the esophageal wall; and for multiple treatment endoscopies. Aqua RFVA System (AquaMedical, Santa Anna, CA) is a novel ablation system that generates vapor/steam at 100°C using an RF electrode at the catheter tip to ablate tissue. It has been designed to address many of the limitations of current ablation techniques. We aimed to develop dosimetry using in-vitro lean-beef, acute and subacute porcine models and test the feasibility, safety and efficacy in patients with flat dysplastic BE.
Aims & Methods: Aqua system includes an RFVA generator (60 W), a 7Fr RFVA catheter and a syringe with saline. The catheter is passed through the biopsy channel of a standard endoscope with distal attachment cap and is used to create focal (∼1 cm2) ablations in the esophagus. The RFVA system was tested in three successive phases; a benchtop lean-beef model; a porcine study (n = 6); and an ongoing, FIH study (n = 15). In the lean-beef and porcine experiment, ablation depth using the RF-VA system at different dosages (dose = duration of vapor delivered in seconds) was compared with ablation depth for focal RFA (1–4 × 12 J/cm2) at acute (0 h) and subacute (48 h) setting. Based on these results, two doses were selected for further testing in the FIH study, including patients with flat dysplastic BE. Per-patient, 4 ablations (2/dose) were applied at 1 cm distance. Patients were followed post-ablation and symptoms and AEs were recorded. Repeat EGD with histology was performed after 6–8 weeks to assess the rate of conversion to squamous epithelium.
Results: None of the treatments in all study-phases resulted in any complications. As expected, in the lean-beef model (total 10 ablations/modality/dose), increasing doses of RFVA and increasing numbers of RFA applications both resulted in increasing depth of ablation. The range of ablation depth with 2–5 s Vapor (0.75 mm–1.5 mm) was comparable to 1–4 × RFA applications (0.58 mm–1.5 mm). In acute porcine evaluation (total 42 Vapor ablations), no differences were found for all doses and treatments tested (3/5 s Vapor and 1–2 × RFA). However, as the treatment effect evolved over the following 48 (total 60 Vapor ablations), a 3 s RFVA was comparable to 2 × RFA, whereas 5 s RFVA produced slightly deeper ablation. We selected a conservative 1 s and 3 s dose for human testing. In this ongoing FIH phase, a total of 42 RFVA were technically successful applied in 12 patients. No adverse events occurred and the procedure was well tolerated, with pain scores ranging from 0–1 out of 10 during 14 days post treatment. Follow-up endoscopy was performed in 6 patients with 24 ablations and showed a median squamous conversion rate for the 1 s and 3 s ablations of 73% (IQR 0–100) and 98% (IQR 0–100), respectively. Histologic analysis from all biopsies obtained from endoscopically eradicated areas, confirmed normal squamous epithelium without buried BE glands.
Conclusion: In this 3-phase study with lean-beef, porcine and the first in-human application, the Aqua RFVA system was safe for esophageal ablation and successfully converted dysplastic BE into squamous epithelium. These preliminary yet promising results warrant further testing with RFVA devices.
Disclosure: Nothing to disclose.
LB10 CLINICAL IMPACT OF ENDOSCOPIC ULTRASOUND-GUIDED THROUGH-THE-NEEDLE MICROBIOPSIES IN PATIENTS WITH PANCREATIC CYSTS – A PROSPECTIVE SINGLE-CENTER STUDY
B. Kovacevic1, P. Klausen1, C. Vestrup Rift2, A. Toxværd3, H. Grossjohann4, J.G. Karstensen5, L. Brink1, H.H. Al-Hashemi1, E. Kalaitzakis1, J. Storkholm4, C.P. Hansen4, J.P. Hasselby2, P. Vilmann1
1Copenhagen University Hospital Herlev and Gentofte, Gastro Unit, Division of Endoscopy, Herlev, Denmark
2Copenhagen University Hospital Rigshospitalet, Department of Pathology, Copenhagen, Denmark
3Copenhagen University Hospital Herlev and Gentofte, Department of Pathology, Herlev, Denmark
4Copenhagen University Hospital Rigshospitalet, Department of Gastrointestinal Surgery, Copenhagen, Denmark
5Hvidovre Hospital, University of Copenhagen, Gastro Unit, Pancreatitis Centre East, Hvidovre, Denmark
Contact E-Mail Address:bojan.k@dadlnet.dk
Introduction: Previously, results of EUS-guided through-the-needle biopsies (TTNBs) from pancreatic cystic lesions (PCLs) have been reported in several, mainly retrospective studies.1-5 While the technique is associated with high technical success and diagnostic yield, there is a lack of evidence concerning its added diagnostic value. Furthermore, there is conflicting evidence regarding the rate of adverse events, the most common of which being acute pancreatitis.
Aims & Methods: The primary aim of this prospective single-center study was to estimate the clinical impact of TTNBs. Between February 2018 and August 2019, we included consecutive patients presenting with a PCL of 15 mm or above referred for EUS examination. Technical success was defined as successful puncture of the lesion with an FNA-needle and acquisition of at least one macroscopically visible biopsy, whereas diagnostic yield was defined as the proportion of the patients where a histological diagnosis was established. Cytological samples were rated according to Papanicolaou Society of Cytopathology guidelines,6 ROSE was not performed. Adverse events were defined according to current ASGE lexicon.7 Demographic and lesion data, together with procedural variables of interest were recorded. Logistic regression and appropriate univariate analyses were performed exploring possible predictors of adverse events.
Results: One hundred and one patients were included. Fifty-four of the patients (53.5%) were female, and mean age was 67.9 (SD 9.9). High-risk stigmata were observed in three patients (3.0%), whereas 53 patients (52.5%) had at least one worrisome feature. Technical success was high (n = 95, 94.1%). Diagnostic yield of TTNB was higher compared with FNA-cytology, 77.1% and 31.4% respectively (p < 0.001). Procedural data and adverse events are presented in Table 1. Adverse event rate did not change significantly following introduction of aggressive perioperative hydration with Ringer lactate and rectal NSAIDs (17.6% vs. 8.3%, p = 0.366). Statistical analyses did not reveal any procedural or demographic variables to be associated with adverse events. Additional diagnostic yield of TTNB, leading to change in clinical management, was 11.9%. Of these, a diagnosis of SCN was made in 10 cases, leading to discontinuation of follow-up. In the remaining two cases, a diagnosis of MCN and IPMN was made in spite of low CEA concentration in cyst fluid (0 and 14 µg/L respectively). Thirteen patients underwent surgical resection, and the concordance rate between TTNB and surgical specimen diagnosis was 89%.
Conclusion: TTNBs had an additional diagnostic yield of 11.9% compared with FNA-cytology and cross-sectional imaging, resulting in important change of clinical management. However, the procedure seems to be associated with severe adverse events and a substantial overall adverse event rate of 9.9%. Further studies will show the proper indications of TTNB and in which subgroup of patients the clinical implication will outweigh the risk.
Disclosure: Nothing to disclose.
Procedural data and adverse events
Lesion size in mm, median (IQR)
25.0 (20.0–31.0)
Total procedural time in min, mean (SD)
23.4 (8.2)
Total intracystic needle time in min, mean (SD)
9.4 (5.0)
Number of biopsies acquired, median (IQR)
2 (2–3)
Length of puncture path in mm, mean (SD)
6.7 (3.9)
Postoperative adverse events by severity, n (%)
Total
10 (9.9)
Mild
4 (4.0)
Moderate
3 (3.0)
Severe
2 (2.0)
Death
1 (1.0)
Postoperative adverse events by diagnosis, n (%)
Acute pancreatitis
9 (8.9)
Admission for observation due to bleeding and free fluid in the lesser sac
1 (1.0)
References
KovacevicBKlausenPHasselbyJP, et al.A novel endoscopic ultrasound-guided through-the-needle microbiopsy procedure improves diagnosis of pancreatic cystic lesions. Endoscopy2018; 50: 1105–1111.YangDSamarasenaJBJamilLH, et al.Endoscopic ultrasound-guided through-the-needle microforceps biopsy in the evaluation of pancreatic cystic lesions: a multicenter study. Endosc Int Open2018; 6: E1423–e1430.YangDTrindadeAJYachimskiP, et al.Histologic analysis of endoscopic ultrasound-guided through the needle microforceps biopsies accurately identifies mucinous pancreas cysts. Clin Gastroenterol Hepatol2019; 17: 1587–1596.BarresiLCrinoSFFabbriC, et al.Endoscopic ultrasound-through-the-needle biopsy in pancreatic cystic lesions: A multicenter study. Dig Endosc2018; 30: 760–770.Crino SF, Bernardoni L, Brozzi L et al. Association between macroscopically visible tissue samples and diagnostic accuracy of EUS-guided through-the-needle microforceps biopsy sampling of pancreatic cystic lesions. Gastrointest Endosc 2019, DOI: 10.1016/j.gie.2019.05.009.PitmanMBCentenoBAAliSZ, et al.Standardized terminology and nomenclature for pancreatobiliary cytology: The Papanicolaou Society of Cytopathology Guidelines. Cytojournal2014; 11: 3–3.CottonPBEisenGMAabakkenL, et al.A lexicon for endoscopic adverse events: report of an ASGE workshop. Gastrointest Endosc2010; 71: 446–454.
LB11 SINGLE-OPERATOR PERORAL CHOLANGIOSCOPY (SPYGLASSDS)-GUIDED BIOPSY VS. ERCP-GUIDED BRUSHING FOR INDETERMINATE BILIARY STRICTURES – A PROSPECTIVE, RANDOMIZED MULTICENTER TRIAL
C. Gerges1, T. Beyna2, R.S.Y. Tang3, F. Bahin4, J.Y.W. Lau5, E.J.M. Van Geenen6, H. Neuhaus7, D.N. Reddy8, M. Ramchandani9
5Hongkong Prince of Wales Hospital, Hongkong, China
6UMC St Radboud, Nijmegen, Netherlands
7Evangelisches Krankenhaus Düsseldorf, Dpt. of Internal Medicine, Düsseldorf, Germany
8Asian Inst. of Gastroenterology – Gastroenterology, Asian Inst. of Gastroenterology; Hyderabad/IN, Gastroenterology, Hyderabad, India
9Asian Institute of Gastroenterology, Gastroenterology, Hyderabad, India
Contact E-Mail Address:gerges.christian@gmail.com
Introduction: Accurate diagnosis of indeterminate biliary strictures is challenging but important for patient prognostication and further management. Biopsy under direct cholangioscopic vision might be superior to standard ERCP techniques such as brushing or biopsy. Our aim was to investigate whether SOC compared with standard ERCP work-up improves the diagnostic yield in patients with indeterminate biliary strictures.
Aims & Methods: Patients with an indeterminate biliary stricture on the basis of MRCP were randomized to standard ERCP visualization with tissue brushing (Control Arm, CA) or single-operator cholangioscopy (SOC) visualization and SOC-guided biopsy (Study Arm, SA). This was a prospective international multicenter trial with a procedure-blinded pathologist.
Results: 61 patients in three tertiary referral centers were consecutively screened and enrolled from May 2017 to December 2018. The study was ended after reaching the primary endpoint and ended at 6 months follow-up of the last enrolled patient. 32 were randomized to SA and 29 to CA. Demographic and baseline characteristics were equally distributed in both arms.
In the CA 44.8% completed the study without malignancy and 48.2% with malignancy. In the SA 25% completed the study without malignancy and 68.8% with malignancy.
Diagnostic Accuracy of Histopathology: Tissue samples could be obtained in all cases at first evaluation. In the SA one sample was inadequate for histological analysis resulting in a yield of SOC on first sample of 96.8%. In the CA brushings were adequate for cytological analysis in all patients. In one participant in the CA the proceduralist suspected a stone as a reason for the stricture with no wire passage being possible. In this case SOC was used and visualized a stone which was not seen on prior imaging and successfully treated with SOC-guided electrohydraulic lithotripsy. No brushings were performed in this case. First sample sensitivity of SOC guided biopsies were significantly higher than ERCP guided brushing (SA 68.2% vs. CA 21.4%, p = < 0.01). All other parameters such as specificity, positive predictive value, negative predictive value and overall accuracy showed no significant difference.
Impact of visual impression: Only one lesion in the SA could not be visualized. In approximately half of the cases in both arms the visual impression had an impact on the patient management (CA 50% vs. SA 56.7%; p = 0.62). The sensitivity (SA 95.5% vs. CA 64.3%; p = 0.01), overall accuracy (OA) (SA 87.1% vs. CA 62.1%, p = 0.03) and PPV (SA 100% vs. CA 81.8%, p = 0.04) were significantly higher in the SA compared with the CA, while specificity and NPV showed no significant difference (Table 4).
Adverse Events: Mild adverse events occurred in three patients in the CA and in two patients in the SA, and could be successfully treated conservatively.
Conclusion: In conclusion, SOC was shown to be safe and effective with a higher diagnostic sensitivity compared with standard ERCP and brushing for the diagnosis of indeterminate biliary strictures. The visual impression was shown to have a pertinent impact on patient management and should be an essential part of determining the correct diagnosis. SOC should be considered as a viable diagnostic alternative to conventional ERCP for indeterminate biliary strictures with the potential to change patient management.
Disclosure: Nothing to disclose.
References
RamchandaniMReddyDNLakhtakiaS, et al.Per oral cholangiopancreatoscopy in pancreatico biliary diseases – expert consensus statements. World J Gastroenterol2015; 21: 4722–4734.NavaneethanUNjeiBLourdusamyVKonjetiRVargoJJParsiMA. Comparative effectiveness of biliary brush cytology and intraductal biopsy for detection of malignant biliary strictures: A systematic review and meta-analysis. Gastrointest Endosc2015; 81: 168–176.DraganovPVChauhanSWaghMS, et al.Diagnostic accuracy of conventional and cholangioscopy-guided sampling of indeterminate biliary lesions at the time of ERCP: A prospective, long-term follow-up study. Gastrointest Endosc2012; 75: 347–353.RamchandaniMReddyDNGuptaR, et al.Role of single-operator peroral cholangioscopy in the diagnosis of indeterminate biliary lesions: A single-center, prospective study. Gastrointest Endosc2011; 74: 511–519.ChenYKParsiMABinmoellerKF, et al.Single-operator cholangioscopy in patients requiring evaluation of bile duct disease or therapy of biliary stones (with videos). Gastrointest Endosc2011; 74: 805–814.
LB12 A SINGLE-USE DUODENOSCOPE FOR ENDOSCOPIC RETROGRADE CHOLANGIOPANCREATOGRAPHY – EARLY RESULTS FROM A CLINICAL CASE SERIES
V.R. Muthusamy1, M.J. Bruno2, R. Kozarek3, B. Petersen4, D. Pleskow5, D. Sejpal6, A. Slivka7, J. Peetermans8, M. Rousseau8, G. Tirrell8, A. Ross3
1University of California, Los Angeles, Vatche and Tamar Manoukian Division of Digestive Diseases, Los Angeles, United States
2University Medical Center Rotterdam, Department of Gastroenterology & Hepatology, Rotterdam, Netherlands
3Virginia Mason Medical Center, Department of Gastroenterology, Digestive Disease Institute, Seattle, United States
4Mayo Clinic, Division of Gastroenterology and Hepatology, Rochester, United States
5Beth Israel Deaconess Medical Center, Division of Gastroenterology, Boston, United States
6Zucker School of Medicine at Hofstra/Northwell, Division of Gastroenterology, Manhasset, United States
7University of Pittsburgh Medical Center, Department of Gastroenterology, Hepatology, and Nutrition, Pittsburgh, United States
Introduction: Patients who undergo endoscopic retrograde cholangiopancreatography (ERCP) may be at risk of infection due to cross-contamination associated with duodenoscope reuse. Guidelines recommend multiple interventions for reprocessing techniques and compliance.1,2 Because high-level disinfection and sterilization may fail,3,4 improved guideline compliance cannot guarantee prevention of exogenous duodenoscope-based transmission of infection.5 Employing single-use duodenoscopes is an alternative strategy to avoid contamination associated with endoscope reuse. A new single-use duodenoscope recently showed comparable performance to three models of reusable duodenoscopes in an ERCP bench simulation study. Performance of the new device should be tested clinically.
Aims & Methods: This Ethics Committee-approved study aimed to assess the safety and feasibility of ERCP using the first-generation EXALT Model D single-use duodenoscope (Boston Scientific, Marlborough, MA; NCT03701958) in adult patients at six academic medical centers. Seven ERCP experts enrolled consecutive adults without altered anatomy who were scheduled for a clinically indicated ERCP. “Roll-in” testing consisted of navigation to and visualization of the duodenal papilla, followed by ERCP with a reusable duodenoscope. After successful completion of roll-in cases, a separate group of patients was enrolled for the ERCP procedural study. Endpoints were ability to complete the roll-in or ERCP, incidence of crossover from single-use duodenoscope to reusable duodenoscope, procedure-related serious adverse events assessed at 72 hours and 7 days, and endoscopist ratings of satisfaction with the single-use duodenoscope.
Results: Seventy-three patients participated as roll-in (n = 13) or ERCP (n = 60) cases in a consecutive case series in April and May 2019. Of the 60 patients who had an ERCP, 37 (61.7%) were male, mean age was 64.0, 44 (73.3%) had a prior ERCP and 43 (71.7%) had a known prior sphincterotomy. American Society for Gastrointestinal Endoscopy (ASGE) procedural complexity scores were Grade 1 (least complex) for seven (11.7%), Grade 2 for 26 (43.3%), Grade 3 for 26 (43.3%), and Grade 4 (most complex) for one (1.7%) cases.
All 13 (100%) roll-in cases were completed without crossover to a reusable duodenoscope. All 60 (100%) ERCPs were completed, 58 (97%) using the single-use duodenoscope only, and 2 (3%) with crossover to a reusable duodenoscope. The most common ERCP indications were for management of biliary stent (n = 33, 55.0%), evaluation of biliary stricture (n = 16, 26.7%), and bile duct stone clearance (n = 11, 18.3%). One crossover case required pancreatic endotherapy in which pancreatic duct (PD) cannulation failed with the single-use and a reusable duodenoscope; PD cannulation ultimately succeeded using needle-knife sphincterotomy. In another crossover case, dilation of a tight hilar biliary stricture in a patient with primary sclerosing cholangitis failed with the single-use duodenoscope, then succeeded with a reusable duodenoscope. Among the 73 patients, three (4.1%) experienced mild or moderate post-ERCP pancreatitis, one (1.4%) had a serious postsphincterotomy bleed, and one (1.4%) with walled-off pancreatic necrosis and pre-existing infection worsened after PD stent placement. Median overall satisfaction with the single-use duodenoscope was rated 9.0 (range 1.0–10.0), with ratings ≥7 in 56 (93.3%) ERCPs.
Conclusion: Expert endoscopists completed consecutive ERCPs using the new EXALT single-use duodenoscope in the great majority of cases, including all four ASGE complexity grades.
Disclosure: VRM: consulting fees and research funding from Boston Scientific Corporation and Medtronic, consultant relationships with Medivators and Interpace, honoraria from Ethicon/Torax, and stockholdership in CapsoVision; MJB: consulting and speaker fees and investigator-initiated grants from Boston Scientific Corporation, Cook Medical, Pentax Medical and 3 M, and consultant fees from Mylan; RAK: research support from Boston Scientific Corporation; BTP: consultant/investigator relationship with Boston Scientific Corporation, consultant fees from Olympus America, Advanced Steriliz Products and GI Medical; DKP: consultant fees from Boston Scientific Corporation, Olympus, Fuji and Nine Point Medical and Medtronic; DVS: consulting/research relationship with Boston Scientific Corporation, consultant fees from Olympus; AS: research funding from Boston Scientific Corporation and Olympus; JAP, MJR and GPT are full-time employees of Boston Scientific Corporation; ASR: consulting fees and research funding from Boston Scientific Corporation.
References
Reprocessing Guideline Task Force, PetersenBTCohenJ, et al.Multisociety guideline on reprocessing flexible GI endoscopes: 2016 update. Gastrointest Endosc2017; 85: 282–294 e1.BeilenhoffUBieringHBlumR, et al.Reprocessing of flexible endoscopes and endoscopic accessories used in gastrointestinal endoscopy: Position Statement of the European Society of Gastrointestinal Endoscopy (ESGE) and European Society of Gastroenterology Nurses and Associates (ESGENA) – Update 2018. Endoscopy2018; 50: 1205–1234.RossASBaligaCVermaPDuchinJGluckM. A quarantine process for the resolution of duodenoscope-associated transmission of multidrug-resistant Escherichia coli. Gastrointest Endosc2015; 82: 477–483.NaryzhnyISilasDChiK. Impact of ethylene oxide gas sterilization of duodenoscopes after a carbapenem-resistant Enterobacteriaceae outbreak. Gastrointest Endosc2016; 84: 259–262.HutflessSMKallooAN. Commentary on the 2016 Multi-Society Task Force Endoscope Reprocessing Guidelines. Gastroenterology2017; 152: 494–496.
LB13 COLONOSCOPY WITH EMBEDDED DEEP LEARNING COMPUTER-AIDED DETECTION SYSTEM IMPROVES ADENOMA DETECTION WITHOUT INCREASING PHYSICIAN FATIGUE: A PROSPECTIVE RANDOMIZED STUDY
P. Wang1, G. Zhou1, J.R. Glissen Brown2, T.M. Berzin2, X. Liu1, L. Li1, W. Liu1, X. Xiao1, Z. Chen1, Z. Zhang1, C. Zhou1, L. Lei1, F. Xiong1, Y. Song1, P. Liu1
1Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, Chengdu, China
2Beth Israel Deaconess Medical Center and Harvard Medical School, Center for Advanced Endoscopy, Boston, United States
Contact E-Mail Address:wangpuhuaxi@qq.com
Introduction: Computer-aided detection (CADe) of colon polyps has been demonstrated to improve colon polyp and adenoma detection during colonoscopy by alarming the location of polyps on a parallel monitor simultaneously. While the video flow on the two monitors are duplicate, the aim of this study was to investigate whether embedding this CADe system into regular colonoscopy could increase polyp and adenoma detection and without increasing physician fatigue level.
Aims & Methods: Consecutive patients presenting for diagnostic and screening colonoscopies were prospectively randomized to undergo routine colonoscopy with or without the assistance of the previously validated real-time polyp detection CADe system which provides a simultaneous visual notice and sound alarm when a polyp was detected. The fatigue level was evaluated from score 0–10 by the performing endoscopists after each colonoscopy procedure. The main outcome was adenoma detection rate (ADR).
Results: Out of 790 patients analyzed, 397 were randomized to routine colonoscopy (control group), and 393 to a colonoscopy with computer-aided diagnosis (CADe group). A total of 625 polyps, 304 adenomas and 12 serrated adenomas were detected. The polyp detection rate (PDR) of the control and CAD groups were 33.25% and 47.07% respectively (OR = 1.786, 95% CI 1.339–2.381, P < 0.001). The adenoma detection rates (ADR) were 20.91% and 29.01%, respectively (OR = 1.56, 95% CI 1.129–2.158, P = 0.009). The average number of polyps per colonoscopy (PPC) were 0.51 and 1.07, respectively (Change Folds = 2.09, 95% CI 1.764–2.464, P < 0.001). The average number of adenomas per colonoscopy (APC) were 0.29 and 0.48, respectively (Change Folds = 1.72, 95% CI 1.367–2.159, P < 0.001). The improvement was mainly due to non-advanced diminutive adenomas, serrated adenoma and hyperplastic polyps. There was a total of 29 false alarms and no missed polyp by the automatic polyp detection. The fatigue score for each procedure was 3.28 vs. 3.40 for routine and CADe group, P = 0.357.
Conclusion: With negligible latency, the real-time CADe embedded into regular colonoscopy could lead to a similar improvement in ADR and PDR without increasing the fatigue level during colonoscopy, which indicates that the embedding of low-latency and high-performance CADe system is an effective quality assurance of colonoscopy.
Disclosure: Tyler Berzin has received consulting fees from Shanghai Wision AI Co., Ltd. The CADe system (EndoScreener) was developed by Shanghai Wision AI Co., Ltd. The system was provided free of charge for the purpose of this study. Employees in the company were not involved in the clinical trial in any way, including in study design, statistical analysis or manuscript writing.
Main outcomes
Routine Colonoscopy (n = 397)
CAD Colonoscopy (n = 393)
P-value**
FC/OR
95% CI
PDR
0.3325
0.4707
<0.001
1.786*
1.339–2.381
ADR
0.2091
0.2901
0.009
1.546*
1.116–2.141
Mean number of detected polyp
0.5139
1.0712
<0.001
2.085#
1.764–2.464
Mean number of detected adenoma
0.2922
0.4784
<0.001
1.637#
1.299–2.063
Fatigue level
3.2821
3.4020
0.357
1.037*
0.960–1.119
PDR, polyp detection rate. ADR, adenoma detection rate. *OR, odds ratio. #FC, fold change. **P-value from χ2 test (or Fisher’s exact test, as appropriate) or t-test.
LB14 LINKED COLOUR IMAGING VERSUS WHITE-LIGHT COLONOSCOPY FOR ADENOMA DETECTION: A MULTICENTRE, RANDOMISED, CONTROLLED TRIAL IN A FIT-BASED COLORECTAL CANCER SCREENING PROGRAMME
S. Paggi1, F. Radaelli2, C. Senore3, R. Maselli4, A. Amato2, G. Andrisani5, F Di Matteo5, S. Grillo6, G. Sereni7, P. Cecinato8, R. Sassatelli9, G. Manfredi10, S. Alicente10, P. Pallini11, L. Milan12, D. Canova12, E. Buscarini10, E. Rondonotti13, A. Repici14, C. Hassan15
1Gastroenterology Unit, Ospedale Valduce, Como, Italy, Como, Italy
2Valduce Hospital, Como, Italy
3AO Città della Salute e della Scienza, CPO Piemonte, Torino, Italy
4Humanitas Clinical and Research Hospital, Rozzano, Italy
5Digestive Endoscopy Unit, Campus Bio-Medico, Rome, Italy, Rome, Italy
6Azienda USL-IRCCS Reggio Emilia, Gastroenterology, PARMA, Italy
7Azienda USL-IRCCS Reggio Emilia, Reggio Emilia, Italy, Reggio Emilia, Italy
8Azienda USL-IRCCS Reggio Emilia, Reggio Emilia, Italy
9Azilenda USL, IRCC Reggio Emilia, Reggio Emilia, Italy
12Azienda ULSS n 8 Berica – Vicenza, Vicenza, Italy
13Ospedale Valduce Gastroenterology Unit, Dept. of Gastroenterology, Como, Italy
14Ist. Clinico Humanitas Rozzano Dept. of Gastroenterology, Dept. of Gastroenterology, Milano, Italy
15Nuovo Regina Margherita Hospital, Gastroenterology, italy, Italy
Contact E-Mail Address:silviapagg@gmail.com
Introduction: Linked-Colour Imaging (LCI), a new image-enhanced technology that emphasizes contrast in mucosal colour, has been demonstrated to substantially reduce the miss rate of polyps as compared with standard White-Light (WL) in tandem colonoscopy studies. Whether LCI increases adenoma detection rate (ADR) is still uncertain.
Aims & Methods: Consecutive subjects undergoing colonoscopy following a positive faecal immunochemical test (FIT) in the context of regional population-based colorectal cancer screening programmes were randomised (ratio 1:1) to undergo colonoscopy with either LCI or WL. Insertion and withdrawal phase of colonoscopy were both carried out using the same assigned light. Experienced endoscopists from seven Italian centres participated in the study. Randomisation was stratified by gender, age group and screening round, using a randomised blocks design. The planned sample size (300 subjects in each arm) could allow for an 80% power to detect as statistical significant (α = 0.05; two-sided test) a 11.5% and 10.5% absolute increase in the detection rate of adenomas and advanced adenomas. The primary outcome measure was the proportion of patients with at least one adenoma (ADR).
Results: Of 704 eligible subjects, 649 were randomised to either LCI (n = 326) or WL (n = 323) colonoscopy and included in the analysis. Overall, 48.9% of patients were male, the mean age ± SD was 60.8 ± 7.3 years, and 32.5% were at first FIT round, with no differences between the two study arms. The ADR was higher in the LCI group (185/ 326 patients, 56.5%) than in the WL group (151/323 patients, 43.5%) (p = 0.047; RR for LCI: 1.22, 95%CI 1.03 to 1.43). The proportion of patients with advanced adenomas and sessile serrated polyps was 26.1% and 8.7% in the LCI arm, and 22.0% and 5.0% in the WL arm, respectively (p = NS for both comparisons).
Multivariate analysis showed that factors independently influencing ADR were the use of LCI, male gender, age greater than 60 years, and adequate (Boston bowel preparation scale ≥6) bowel preparation (Table 1). At per-polyp analysis, the mean number of adenomas per patient was 1.12 ± 1.59 and 0.98 ± 1.47 in the LCI and WL group, respectively (p = NS).
Conclusion: In FIT-positive patients undergoing screening colonoscopy, the routine use of LCI led to a significant increase in ADR.
Disclosure: Nothing to disclose.
Factors associated with ADR at multivariable analysis
Variables
Odds ratio
95% CI
Women (vs. Men)
0.47
0.33–0.66
Age: 60–74 yrs (vs. 50–59 yrs)
1.77
1.23–2.53
FIT round: > 1 (vs. first round)
0.70
0.47–1.04
BBPS ≥6 (vs. <6)
3.35
1.03–10.85
Withdrawal times ≥6 min (vs. <6 min)
1.36
0.65–2.82
Study arm: LCI (vs. WL)
1.53
1.09–2.15
14:00–15:30 / C2
From eosinophilic oesophagitis to novel obesity therapies
LB15 EFFICACY AND SAFETY OF AK002 IN ADULT PATIENTS WITH ACTIVE EOSINOPHILIC GASTRITIS AND/OR EOSINOPHILIC GASTROENTERITIS: PRIMARY RESULTS FROM A RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED PHASE 2 TRIAL (ENIGMA STUDY)
E. Dellon1, K. Peterson2, J. Murray3, G. Falk4, N. Gonsalves5, M. Chehade6, J. Leung7, R. Genta8, M. Rothenberg9, P. Khoury10, A. Bledsoe3, C. Shaw11, H. Rasmussen11, B. Singh11, A. Chang11, A. Kamboj11, I. Hirano12
1University of North Carolina at Chapel Hill, Chapel Hill, United States
2University of Utah School of Medicine, Salt Lake City, United States
3Mayo Clinic, Rochester, Rochester, United States
4University of Pennsylvania Perelman School of Medicine, Pennsylvania, United States
5Northwestern University School of Medicine, Chicago, United States
6Icahn School of Medicine at Mount Sinai, New York, United States
7Tufts Medical Center, Boston, United States
8University of Texas (UT) Southwestern, Dallas, United States
9Cincinnati Children's Hospital Medical Center, Cincinnati, United States
10National Institutes of Health / NIAID, Bethesda, United States
11Allakos, Redwood City, United States
12Northwestern University, Division of Gastroenterology, Chicago, United States
Contact E-Mail Address:murray.joseph@mayo.edu
Introduction: Eosinophilic gastrointestinal (GI) disorders (EGIDs) such as eosinophilic gastritis (EG), gastroenteritis (EGE) and oesophagitis (EoE), are caused by accumulation and activation of eosinophils (eos) and mast cells (MC), resulting in chronic debilitating symptoms (e.g. abdominal pain, nausea, diarrhea, bloating, abdominal cramping, early satiety, vomiting, and dysphagia). There is no FDA-approved treatment for EGIDs and current treatment options are limited. AK002, an anti-siglec-8 antibody, depletes eos and inhibits MC activity, providing a novel targeted therapy for EGID patients.
Aims & Methods: ENIGMA was a randomized, double-blind, placebo-controlled phase 2 clinical trial aimed to assess the efficacy and safety of AK002 in adult patients with EG/EGE. Biopsy-confirmed EG/EGE patients with active moderate to severe symptoms were randomized to AK002 (low dose 0.3–1.0 mg/kg [LD] or high dose 0.3–3.0 mg/kg [HD]) or placebo (PBO) cohorts. Symptoms were assessed using a validated, proprietary, daily EG/EGE patient-reported outcome questionnaire to generate a total symptom score (TSS). The primary endpoint was mean percent change in GI tissue eos counts from baseline (BL). Secondary endpoints were the proportion of treatment responders (defined as patients with both >75% decrease in tissue eos and >30% improvement in TSS), and mean percent change in TSS from BL. IRB approval was obtained.
Results: 59 patients were evaluable for efficacy (n = 20 in HD; n = 19 in LD; n = 20 in PBO). BL characteristics were balanced between groups. For the primary endpoint, the AK002 groups had an overall 95% mean reduction of tissue eos relative to BL compared with a 10% mean increase in PBO (p < 0.0001; Table 1). Tissue eos depletion to ≤ 6 eos/hpf was seen in 37 (95%) AK002 patients. There was significant improvement in TSS scores with AK002 compared with PBO (p = 0.0012). Among all AK002 patients, 69% were treatment responders compared with 5% of PBO patients (p = 0.0008). Among EG/EGE patients with concomitant EoE, significant histologic and substantial symptomatic improvements were reported with AK002 compared with PBO. The most common adverse events (AE) reported for AK002 were mild to moderate infusion related reactions (IRR), most common at the first infusion only. Treatment-emergent serious AEs were similar between AK002 and PBO groups. There was one drug-related serious AE, an IRR that resolved within 24 hours without sequelae.
Conclusion: In the first RCT in EG/EGE patients, AK002 treatment resulted in depletion of GI tissue eos and significant improvement of EG/EGE symptoms compared with PBO. In addition, AK002 treatment resulted in histologic and symptomatic improvements in patients with concomitant EoE. AK002 was also well tolerated and is a promising candidate for targeted treatment of EGIDs.
Disclosure: AK002 is an investigational drug candidate that is not FDA or EMA approved. Dr. Dellon – Consultant, Grant/Research Support, Advisory Board Member: Allakos, Inc. Dr. Peterson – Grant/Research Support: Allakos, Inc. Dr. Murray – Grant/Research Support: Allakos, Inc. Dr. Falk – Grant/Research Support: Allakos, Inc. Dr. Gonsalves – Grant/Research Support, Advisory Board Member: Allakos, Inc. Dr. Chehade – Grant/Research Support, Advisory Board Member: Allakos, Inc. Dr. Leung – Grant/Research Support: Allakos, Inc. Dr. Genta – Consultant, Grant/Research Support, Advisory Board Member: Allakos, Inc. Dr. Rothenberg – Grant/Research Support: Allakos, Inc. Dr. Khoury – Nothing to disclose Dr. Bledsoe – Nothing to disclose Camilla Shaw – Employee: Allakos, Inc. Dr. Rasmussen – Employee: Allakos, Inc. Dr. Singh – Employee: Allakos, Inc. Alan Chang – Employee: Allakos, Inc. Dr. Kamboj – Employee: Allakos, Inc. Dr. Hirano – Consultant, Grant/Research Support, Advisory Board Member: Allakos, Inc.
Primary and Secondary Endpoints
AK002 Dose Groups
Endpoint
High Dose (n = 20)
Low Dose (n = 19)
Combined (n = 39)
Placebo (n = 20)
Primary – Tissue Eosinophils
Mean % Δ from BL to End of Study
–97%
–92%
–95%
+10%
p-value
<0.0001
<0.0001
<0.0001
–
Secondary – Treatment Responders
% Pts with Eos Δ >–75% & TSS Δ >–30%)
70%
68%
69%
5%
p-value
0.0009
0.0019
0.0008
–
Secondary – Total Symptom Score
Mean % Δ from BL to End of Study
–58%
–49%
–53%
–24%
p-value
0.0012
0.0150
0.0012
–
LB16 SAFETY AND EFFICACY OF 4 DOSAGE REGIMENS OF APT-1011: RESULTS FROM A PHASE 2B RANDOMIZED DOUBLE-BLIND PLACEBO-CONTROLLED TRIAL TO SELECT THE BEST DOSAGE REGIMEN FOR FURTHER DEVELOPMENT. (CLINICALTRIALS.GOV NCT03191864; EUDRACT NUMBER: 2016-004749-10)
E.S. Dellon1, A. Lucendo Villarin2, C. Schlag3, A. Schoepfer4, K. Knoop5, P. Richardson5, G. Falk6, G. Eagle Taran7, G.M. Comer7, I. Hirano8
1University of North Carolina School of Medicine, Chapel Hill, United States
2Hospital General de Tomelloso, Aparato Digestivo, Tomelloso, Spain
3Technische Universität München, II. Medizinsiche Klinik, München, Germany
4Centre Hospitalier Universitaire Vaudois, Gastroenterology and Hepatology, Lausanne, Switzerland
5Adare Pharmaceuticals, Clinical Operations, Lawrence Township, United States
6University of Pennsylvania Perelman School of Medicine, Philadelphia, United States
7Adare Pharmaceuticals, Lawrence Township, United States
8Northwestern University, Division of Gastroenterology, Chicago, United States
Contact E-Mail Address:mk8@grupo1492.com
Introduction: Eosinophilic esophagitis (EoE) is an increasingly prevalent atopic condition characterized by eosinophilia of the esophageal mucosa and symptoms of esophageal dysfunction. Fluticasone use has been reported for EoE with formulations not designated for esophageal delivery. APT-1011, a novel fluticasone propionate orally disintegrating tablet formulation, is under investigation for the treatment of EoE.
Aims & Methods: To evaluate the safety and efficacy of four dosing regimens of APT-1011 and select the dosing regimen with the best benefit to risk profile for further development.
Methods: The FLUTicasone in EoE (FLUTE) trial was a randomized, double-blind, placebo-controlled, multicenter, dose-ranging study of APT-1011 vs. placebo in adults diagnosed with EoE. Participants were enrolled from 71 sites in six countries in North America and Western Europe from September 2017 to October 2018. Participants were randomized to APT-1011 (1.5 mg HS; 1.5 mg AM and HS; 3 mg HS; or 3 mg QAM and HS) or matching placebo. The primary efficacy endpoint was histologic response at Week 12, as measured by percent of study participants with a peak of ≤6 eosinophils per high power field (eos/HPF) on at least five esophageal biopsies. Secondary endpoints assessed endoscopic activity and symptoms. Safety parameters were also assessed. (Clinicaltrials.gov NCT03191864; EudraCT Number: 2016-004749-10).
Results: At total of 103 patients were enrolled (mean age 38.6; 68.0% male; 97.1% white). An overall response rate of 70% was observed among all APT-1011-treated study participants, compared with 0% of placebo-treated study participants. Among the APT-1011 dose groups, the 1.5 mg BID dose yielded the highest histologic response rate (86.4%) (Table). A total daily dose of 6 mg (3 mg BID) did not provide histologic benefit beyond the 3 mg daily dose (1.5 mg BID or 3 mg HS). Statistically significant improvements in endoscopic appearance were observed with all APT-1011 dosing groups compared with placebo, with the 3 mg HS dosing regimen showing the largest numerical improvement. The most common treatment-emergent adverse events were esophageal 8/85 (9.4%) and oral candidiasis 5/85 (5.9%) in subjects on APT-1011, compared with 0% on placebo. Most events of candidiasis were in the 3 mg BID dosing group (8/20; 40%). No subject experienced adrenal axis suppression based on adrenocorticotropic hormone simulation testing or symptoms of hypercortisolism.
Conclusion: APT-1011 for 12 weeks is highly efficacious to induce histologic remission and is safe and well tolerated in adult patients with EoE. The 3 mg HS dose proved to have the best benefit to risk balance of efficacy and safety, with the added benefit of once daily dosing, for future development. Maintenance therapy for up to 52 Weeks is currently ongoing.
Disclosure: Evan Dellon has received research funding from Adare, Allakos, GSK, Meritage, Miraca, Nutricia, Celgene/Receptos, Regeneron, Shire; and consulting fees from Adare, Aimmune, Alivio, Allakos, AstraZeneca, Banner, Biorasi, Calypso, Celgene/Receptos, Enumeral, EsoCap, Gossamer Bio, GSK, Regeneron, Robarts, Salix, Shire; and educational grant from Allakos, Banner, Holoclara.
Endpoints at 12 Weeks
APT-1011 3 mg BID (N = 20)
APT-1011 3 mg HS (N = 21)
APT-1011 1.5 mg BID (N = 22)
APT-1011 1.5 mg HS (N = 21)
Placebo (N = 19)
Histologic Response Rate
80.0%
66.7%
86.4%
47.6%
0%
EREFs Total Score
Baseline, mean
4.3
5.1
4.5
5.1
5.1
Week 12, mean
−2.2
−3.3
−2.7
−2.4
−0.9
90% confidence interval LS mean difference
(−2.58, −0.55)
(−3.31, −1.33)
(−3.03, −1.04)
(−2.35, −0.32)
1-sided p-value
0.006
<0.001
<0.001
0.016
Global EoE Symptom Score
Baseline, mean
5.8
6.0
5.8
5.6
5.6
Week 12, mean
−1.7
−3.1
−1.6
−3.2
−1.8
90% confidence interval of LS mean difference
(−0.83, 1.80) 1-sided p-value 0.729
(−2.45, 0.11) 1-sided p-value 0.065
(−0.80, 1.74) 1-sided p-value 0.729
(−2.44, 0.22) 1-sided p-value 0.084
Table reference: * Responder is defined as a subject with ≤6 peak eosinophils/high-power field (HPF). EREFs: Eosinophilic Esophagitis Endoscopic Reference Score; EoE: eosinophilic esophagitis; LS: least square; N: number
LB17 MICROBIOTA DYSBIOSIS INDUCES COLORECTAL CANCER (CRC) THROUGH AN EPIGENETIC MECHANISM: EXPERIMENTAL RESULTS IN MICE, AND VALIDATION IN HUMAN PILOT (N = 266) AND PROSPECTIVE (N = 1000) STUDIES
I. Sobhani1, E. Bergsten2, A. Amiot3, S. Couffin4, N. DeAngelis3, S. Rabot5, D. Mestivier6, T. Pedron7, K. Khazaie8, P. Sansonetti9; Oncomix (Microbiota and Cancer) Study Group
1UPEC-APHP-Hopital Henri Mondor-APHP, Gastroenterology, Creteil, France
2Universtite Paris Est Creteil, EC2M-EA7375, Creteil, France
3Henri Mondor Hospital, APHP Dept. of Gastroenterology, Dept. of Gastroenterology, Creteil, France
4UPEC-APHP-Hopital Henri Mondor-APHP, EC2M-EA7375, Creteil, France
5INRA, AgroParisTech, Université Paris-Saclay, Micalis Institute, Jouy en Josas, France
6UPEC, EC2M-EA7375, Creteil, France
7INSERM U1202-Institut Pasteur, Paris, Paris, France
8Mayo Clinic, Surgery, Rochester, United States
9INSERM U1202-Institut PASTEUR, Paris, France
Contact E-Mail Address:iradj.sobhani@aphp.fr
Introduction: Sporadic colorectal cancer (CRC) is a result of complex interactions between the host and its environment. Environmental stressors act by causing host-cell DNA alterations (genetic and epigenetic changes). Interest of microbiota in mass screening for colon cancer has not been prospectively investigated.
Aims & Methods: To (1) investigate the stressor ability of CRC-associated gut dysbiosis as causal agent of host DNA alterations in germ-free animals; (2) identify genetic and epigenetic changes in the colonic mucosa; (3) find out similar alterations in human donors; (4) validate data in human cohorts.
Germ-free mice (approval ethical committee #12/076; n = 185) received fresh fecal samples from nine subjects with normal colonoscopy and nine gender- and age-matched CRC patients, and were monitored (7–14 weeks) after human fecal transfer (FMT). Aberrant crypt foci, luminal microbiota and DNA alterations (whole colonic exome sequencing and levels of gene methylation in tissues and effluents) were monitored following FMT by using Agilent SureSelect technology (HiSeq 2000) and methylchips (Illumina GoldenGate®) procedures. Food uptake, weight of animals and blood routine tests were monitored. Fecal bacteria in animals were characterized (16sRNA) at baseline (human donors) and over time during the follow up. Associations between DNA (genetic, epigenetic) and stool bacteria were established by using appropriate bioinformatic methods. Significant associations were investigated in a human pilot study (n = 266), and a panel of dysbiosis-related methylate genes was validated in asymptomatic (n = 500) and symptomatic (n = 500) individuals undergoing colonoscopy and followed up for 3–5 years. Human fecal microbiota was finally submitted to whole metagenomic sequencing for the identification of bacteria species associated with DNA alteration in the validation prospective (Clinical Trial NCT01270360).
Results: After human FMT, CRC-associated microbiota induced higher numbers of ACF and higher hypermethylated genes in the mice colonic mucosa (vs. healthy controls' microbiota recipients) when no significant difference in DNA mutation was observed in between animal groups. As suggested by these data, in human effluents (blood and stools) and tissues from donors, several gene promoters including SFRP1,2,3, PENK, NPY, ALX4, SEPT9, WIF1 were found hypermethylated in human CRC individuals; the methylation status of these genes was associated with panel of bacteria whose abundances were significantly different versus healthy donors.
In a pilot study (n = 53 CRCs, 123 controls), the blood methylation levels of three genes (Wif1, PENK and NPY) were shown closely associated with dysbiosis. Consequently a cumulative methylation index (CMI) of these genes was constructed. In a prospective validation study (n = 187 CRCs; 813 controls) with at least 3 years follow-up, the CMI of these genes is found significantly higher in CRC patients as compared with controls. The multivariate analysis that included also fecal immunochemical blood test (FIT) in asymptomatic individuals showed CMI as an independent risk factor for CRC diagnosis. Finally, fecal bacterial species in the validation cohort were identified based on their whole genome sequences and panel of pro epigenomic bacteria in stools was established.
Conclusion: CRC-related dysbiosis induces methylation rather than mutation in host genes. Promethylator bacteria are characterized in stools. Their quantification is useful for the identification of individuals with high risk of colon cancer. Promethylator dysbiosis can be used for the evaluation of probiotics in preventing colon cancer.
Disclosure: Nothing to disclose.
LB18 CNP-101 PREVENTS GLUTEN CHALLENGE-INDUCED IMMUNE ACTIVATION IN ADULTS WITH CELIAC DISEASE
C. Kelly1,2, J. Murray3, D.A. Leffler4, A. Bledsoe3, G. Smithson4, J. Podojil5, R. First5, A. Morris5, M. Boyne5, A. Elhofy5, T.T. Wu3, S. Miller5,6
1Beth Israel Deaconess Medical Center, Boston, United States
2Harvard Medical School, Boston, United States
3Mayo Clinic, Rochester, United States
4Takeda Pharmaceuticals International Co., Cambridge, United States
5COUR Pharmaceuticals Development Co., Inc., Northbrook, United States
6Feinberg School of Medicine, Chicago, United States
Contact E-Mail Address:ckelly2@bidmc.harvard.edu
Introduction: In Celiac Disease (CD), gluten triggers an autoimmune process leading to small intestine mucosal injury. There is no approved treatment for CD. CNP-101*(TIMP-GLIA), a gliadin protein within a negatively charged polymer matrix of poly(DL-lactide-coglycolide (PLGA) nanoparticles, is a novel, first-in-class compound designed to induce tolerance to gluten in patients with CD.
Aims & Methods: This Phase 2 double-blind randomized proof-of-concept study was designed to evaluate the efficacy and safety of CNP-101 8 mg/kg versus placebo in celiac patients. Treatments were administered intravenously on Day 1 and Day 8 for prevention of immune activation and small intestinal mucosal injury in adults with well-controlled biopsy-proven CD undergoing gluten challenge. Gluten challenge began 7 days after second treatment administration and included 12 g of gluten per day for 3 days followed by 6 g of gluten per day for 11 days. Endoscopy was performed before treatment and at the end of gluten challenge. The primary endpoint was change from baseline in interferon-gamma (IFN-γ) spot-forming units (SFUs) at Day 6 of gluten challenge using a gliadin-specific enzyme-linked immunospot (ELISpot) assay. Key secondary endpoints were changes from baseline in the villus height to crypt depth ratio (Vh:Cd), intestinal intraepithelial lymphocytes (IELs), peripheral gliadin-specific T-cell proliferation, cytokine secretion, and percentage of gut-homing CD4, CD8, and γδ cells.
Results: A total of 34 patients were randomized and treated; six discontinued due to gluten-related symptoms, and 28 completed the 14-day gluten challenge per protocol. The primary study endpoint was achieved with mean change from baseline in IFN-γ ELISpot of 2.10 with CNP-101 and 17.57 with placebo (p = 0.0056). Mean reduction from baseline in Vh:Cd was 0.18 with CNP-101 and 0.63 with placebo, (p = 0.079). Mean change from baseline in IELs was 28.6 with CNP-101 and 35.0 with placebo (p = 0.289). No patient had clinically significant changes in vital signs, routine clinical labs, or serum cytokines/chemokines, gliadin-specific T-cell proliferation and cytokine secretion. Plasma complement levels were raised in most patients; however, this transient effect was not reflected in associated adverse events. The most frequent AEs observed in CNP-101 treated patients with the corresponding value in parentheses for placebo-treated patients were: nausea 81% (72%), abdominal distension 56% (61%), diarrhea 50% (50%), headache 44% (17%), abdominal pain 38% (28%), abnormal gastrointestinal sounds 38% (39%), vomiting 31% (33%), fatigue 31% (50%), and back pain 31% (0).
Conclusion: In this Phase 2 proof-of-concept study, CNP-101 (TIMP-GLIA) was able to prevent gluten challenge-induced immune activation in adults with CD. To our knowledge, this is the first clinical trial to demonstrate non-autologous induction of antigen-specific immune tolerance in any autoimmune disease.
Disclosure: CK is a consultant to COUR Pharmaceuticals Development Co., Inc., Glutenostics, Immunogenx, Innovate, Takeda, has stock options in Cour Pharma, Glutenostics and is an investigator for Allergan, Innovate, Immunogenx, Takeda. JM is a consultant to Celimmune Amgen actobiotics, Kanyos and Bioniz and has received grants to Mayo Clinic from Immunogenix, DBV technologies, Allakos, Cour Pharma and NIH. RF is a consultant to COUR Pharmaceuticals Development Co., Inc. SM is a consultant to and owns stock in COUR Pharmaceuticals Development Co., Inc. AM was an employee of COUR Pharmaceuticals Development Co., Inc. at the time of the study. JP, AE and MB are employees of COUR Pharmaceuticals Development Co., Inc. DL and GS are employees of Takeda Pharmaceuticals Inc. Co. AB and TTW has no conflicts of interest to declare.
LB19 ASCERTAINMENT OF PANCREATIC CANCER BY LIQUID BIOPSY ASSESSING TWIST1 MRNA LEVELS IN BLOOD
L. Greco1, T. Cavalleri1, F. Gavazzi2, R. Maselli3, S. Galtieri3, G. Carrara2, M. Capretti3, Di Leo, L. Craviotto5, V. Poliani4, P. Preatoni5, P.D. Omodei5, V. Torri6, A. Repici3, A. Zerbi2, A. Malesci5, L. Laghi5
1Humanitas Clinical and Research Center, IRCCS, Dept. of Gastroenterology – Laboratory of Molecular Gastroenterology, Rozzano – MI, Italy
2Humanitas Clinical and Research Center, IRCCS, U.O. Pancreatic Surgery Division, Rozzano – MI, Italy
3Humanitas Clinical and Research Center, IRCCS, Dept. of Gastroenterology, Digestive Endoscopy Unit, Rozzano – MI, Italy
5Humanitas Clinical and Research Center, IRCCS, Dept. of Gastroenterology, Rozzano – MI, Italy
6Laboratory of Clinical Research in Oncology, Istituto di Ricerche Farmacologiche Mario Negri, IRCCS, Milan, Italy
Contact E-Mail Address:luigi.laghi@humanitas.it
Introduction: Delayed diagnosis affects the fate of patients with pancreatic ductal adenocarcinoma (PDAC). In addition, despite the advances in molecular typing, expression signatures exploitable for its diagnosis remain underdeveloped. Surprisingly, epithelial-to-mesenchymal Transition (EMT), the hallmark of PDAC spread in mechanistic models, remains widely untraced in humans.
Aims & Methods: The aim of the study was to assess whether the levels of EMT transcription factors (TF) mRNAs in the blood of PDAC patients are increased, and if so whether they could serve as diagnostic tools.
By quantitative real-time PCR, we assessed the levels (as normalized threshold cycles, ΔCt) of TWIST1, ZEB2, and CDH1 EMT-TF mRNAs in blood samples of 93 PDAC patients and 292 control subjects. Methods are covered by patent EP n.13197367.9.
Results: Circulating mRNA levels of EMT-TFs were higher in patients, with a fold-change of 6.3 times for TWIST1, 10.6 times for ZEB2, and 6.0 times for CDH1 vs. controls. By ΔCt, the median of TWIST1 mRNA was 25.9 in patients vs. 28.3 in controls, that of ZEB2 mRNA 17.6 in patients vs. 22.0 in controls, and that of CDH1 23.6 vs. 26.4 (all p < 0.0001, at rank-sum test). Consistently, the area under the curve (AUC) at receiver operating characteristic curve analysis was 0.94 for TWIST1, 0.88 for ZEB2, and 0.90 for CDH1 mRNAs. By cut-off values, 190 (65%) controls did not show any elevated messenger, a finding never occurring in patients. Conversely, all EMT-TF mRNAs were elevated in 60 (61.8%) patients but only in 10 (3.4%) controls (both p < 0.0001). A regression model for PDAC diagnosis by EMT messengers identified 82 patients, (88.2% sensitivity) and 10 controls (96.6% specificity), with 0.98 AUC. Two-fold cross-validation analysis returned an equal AUC (95% CI, 0.96–0.99). A sub-analysis restricted to 59 patients with stage I–II PDAC who underwent surgery also returned 0.98 AUC, with 86.4% sensitivity (eight false negative).
Conclusion: Our study first demonstrates an enhanced expression of EMT-TF mRNAs in patients with PDAC, and shows that their high levels in blood efficiently discriminate PDAC patients, irrespectively of tumour respectability. These circulating messengers represent a candidate diagnostic tool, deserving further exploration in appropriate clinical settings.
Disclosure: Luigi Laghi is granted as inventor in the EP n.13197367.9.
LB20 A 20-GENE SIGNATURE IN LIVER BIOPSIES OF CHRONIC HEPATITIS B AND C PATIENTS ALLOWS AN ACCURATE PREDICTION OF HCC DEVELOPMENT 8 YEARS BEFORE CLINICAL DIAGNOSIS
S. Van Hees1,2, B. Cuypers3,4, S. Bourgeois2,5, K. De Groot-Kreefft6, D. Sprengers7, G. Robaeys8, P. Meysman3, L. Vonghia1,2, P. Michielsen1,2, S. Francque1,2, R. De Man6, A. Driessen9, A. Boonstra6, K. Laukens3, T. Vanwolleghem1,2,6
1University of Antwerp, Department of Experimental Medicine and Paediatrics, Antwerp, Belgium
2Antwerp University Hospital, Department of Gastroenterology and Hepatology, Antwerp, Belgium
3University of Antwerp, Department of Mathematics and Computer Sciences, Antwerp, Belgium
4Institute of Tropical Medicine, Department of Biomedical Sciences, Antwerp, Belgium
5ZNA Stuivenberg, Department of Gastroenterology and Hepatology, Antwerp, Belgium
6Erasmus Medical Center Rotterdam, Department of Gastroenterology and Hepatology, Rotterdam, Netherlands
7GZA Antwerp, Department of Gastroenterology and Hepatology, Hove, Belgium
8Ziekenhuis Oost-Limburg, Department of Gastroenterology and Hepatology, Genk, Belgium
9University Hospital Antwerp, Department of Pathology, Antwerp, Belgium
Introduction: Hepatocellular carcinoma (HCC) develops in 1–6% of cirrhotic Chronic Hepatitis B (CHB) and C (CHC) patients annually, but an accurate prediction of HCC development remains impossible as of now.
Aims & Methods: In the current study, we explored (1) baseline liver transcriptome profiles of CHB and CHC patients with and without HCC development during long-term follow-up and (2) investigated their potential to predict HCC development. Charts from all CHB and CHC patients from five hepatology clinics were reviewed for the availability of a baseline, pre-antiviral treatment liver biopsy. Of these, 34 were diagnosed with HCC at a median of 8.3 (IQR 4.8–10.1) years after liver biopsy. Cases were split into four subgroups based on infecting virus (HBV/HCV) and cirrhosis status (yes/no) at baseline biopsy (Table 1). Each subgroup of cases was matched for different demographic (e.g. gender and age at biopsy) and clinical (e.g. cirrhosis at biopsy and infecting virus) factors to a group of controls without HCC development during an equal or longer follow-up time. RNA derived from baseline biopsies (total n = 72; Table 1) was subjected to RNA sequencing (RNAseq). Differentially Expressed Genes (DEG; Fold-change >1.5 and q < 0.2) were called in each subgroup (Table 1) and a random forest classifier was trained to predict HCC development.
Results: The total cohort consisted of 72 patients, of whom 34 developed a HCC during follow-up. Despite perfect matching for the clinical and demographic characteristics at baseline, at least 452 genes were differentially expressed between cases with and controls without future HCC development in each subgroup (Table 1). There was little overlap (<10%) in DEG between cirrhotic CHB and CHC patients (Table 1) and non-cirrhotic CHB and CHC patients (Table 1), suggesting that distinct processes leading to HCC oncogenesis may be triggered by these viruses. Among the top 20 up- and down-regulated genes in each subgroup, 40–75% have previously been linked to oncogenesis, underlining the biological relevance. Ingenuity Pathway Analysis showed an enrichment for the “Wnt-β catenin signalling” pathway in the cirrhotic CHB group and the “molecular mechanisms of cancer” pathway in the non-cirrhotic CHC group. These results strongly suggest a genetic imprint for HCC development several years before clinical diagnosis. A random forest classifier tested with leave-one-out-cross-validation was able to predict HCC development with an accuracy of 84.7%, a Negative Predictive Value of 92.1% and a Positive Predictive Value of 75.8% based on the subgroup and baseline expression levels of 20 genes, of whom several have previously been linked to hepatocarcinogenesis.
Conclusion: In this study, we are the first to identify distinct liver transcriptome profiles for patients with and without future HCC development at a median of 8.3 years before the HCC clinical onset. A 20-gene signature allowed us to accurately predict HCC development.
Disclosure: Nothing to disclose.
Overview of the number of cases and controls and the number of DEG detected between cases and controls in each subgroup
Subgroup
Cases (n)
Controls (n)
DEG (n)
Overlapping DEG (n)
Cirrhotic chronic hepatitis B patients
11
11
452
16
Non-cirrhotic chronic hepatitis B patients
7
11
1100
Cirrhotic chronic hepatitis C patients
9
9
2074
212
Non-cirrhotic chronic hepatitis C patients
7
7
2089
LB21 A NOVEL INGESTIBLE INTRALUMINAL GASTROINTESTINAL STIMULATION CAPSULE SYSTEM FOR TREATMENT OF OBESITY – FIRST-IN-HUMAN STUDY
R. Ringel1, T. Naftali2,3, F. Benjaminov2,3, A. Melamud4, Y. Ringel2,3,5
1Columbia University, New York, United States
2Meir Medical Center, Division of Gastroenterology and Hepatology, Kfar Saba, Israel
3Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
4Melcap Systems, Haifa, Israel
5University of North Carolina at Chapel Hill, School of Medicine, Chapel Hill, North Carolina, United States
Contact E-Mail Address:roeyringel@gmail.com
Introduction: Gastric electrical stimulation (GES) has been shown to reduce appetite, food intake and weight, thus leading to recent FDA approval of a surgically implanted gastrostimulator device for treatment of obesity. However, surgically or endoscopically implanting gastrointestinal (GI) stimulating devices can be unnecessarily invasive and expensive. We conducted the first-in-human study to examine the safety, feasibility, and signals of efficacy of a novel ingestible gastric stimulation capsule (IGSC) system for the treatment of obesity. This first-in-human study is an extension of a previous safety and proof-of-concept animal (pigs) study.
Aims & Methods: Six males and females healthy volunteers, age 21 to 65 years, BMI 27 to 35 (overweight and class I obese) were enrolled into a 7-day, prospective, open-label randomized study. Study design included a 3-day screening period, followed by a 4-day treatment period, during which subjects ingested one IGSC on days 1 and 3. The capsules were programmed to apply intra-gastric and/or intestinal electrical stimulation, synchronized with subjects’ meal schedule using the system’s mobile app. Variables of interest for safety included integrity and natural expulsion of the capsule with bowel movement, all reported symptoms and adverse events, and 24 hours holter ECG monitoring. Efficacy measures included satiety and hunger levels, and response to a nutrient drink test. Following the drink test, patients were asked to score their hunger and satiety levels, on a 0–10 visual analog scale using the system’s mobile app, at 30, 60, 120, and 180 minutes. A composite hunger/satiety score was calculated.
Results: All subjects expelled the IGSCs naturally. Seven adverse events (AEs) were reported; all were mild, short-lived, and self-resolving. One event of abdominal discomfort was considered likely related to the study treatment. The other six AEs were unrelated or likely unrelated to the study treatment. No clinically significant abnormal physical findings, laboratory measurements, or heart activities were recorded throughout the course of the study. The IGSC treatment was associated with a decrease in the volume of liquid ingested on the nutrient drink test. Strikingly, four of these individuals consumed an average of 27% less of the nutrient volume after receiving the IGSC treatment when compared with their no-treatment baseline. In addition, the IGSC treatment was associated with a considerable decrease in the hunger/satiety composite score compared with baseline.
Conclusion: Gastric electrical stimulation via the non-invasive, IGSC system proved to be safe, feasible and well tolerated with no serious adverse events and no effect on heart activity. Our results suggest that GI stimulation using IGSC system may (1) reduce food intake as measured by total volume ingested by the nutrient drink test; and (2) reduce appetite as measured by the post nutrient drink hunger/satiety composite scores. This first of its kind IGSC system highlights the potential and provides the rationale for intraluminal GI electrical neuromodulation therapeutic approach. Specifically, the IGSC system could provide a safe, minimally invasive, non-chemical approach for treatment of obesity.
Disclosure: This study was funded by Melcap Systems. YR is consultant for Melcap, AM is an employee of Melcap.
12:30–13:30 / C1
Global burden of gastrointestinal disorders
LB22 GLOBAL, REGIONAL, AND NATIONAL BURDEN OF INFLAMMATORY BOWEL DISEASE AND ITS RISK FACTORS IN 195 COUNTRIES AND TERRITORIES, 1990–2017: A SYSTEMATIC ANALYSIS FOR THE GLOBAL BURDEN OF DISEASE STUDY 2017
S. Alatab1, S.G. Sepanlou1, H. Vahedi1, A.R. Sima1, M.M. Malekzadeh1, A. Sadeghi1, M.A. Dirac2, M. Naghavi2, R. Malekzadeh1
1Digestive Diseases Research Center, Digestive Diseases Research Institute, Shariati Hospital, Tehran University of Medical Sciences, Tehran, Iran (Islamic Republic of)
2Institute for Health Metrics and Evaluation, University of Washington, Seattle, United States, Washington, United States
Introduction: The burden of inflammatory bowel disease (IBD) is rising globally. In this study, we aimed to report the global prevalence, mortality, and overall burden of IBD, based on data from the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD) 2017.
Aims & Methods: We modelled mortality due to IBD using a standard Cause of Death Ensemble model including data mainly from vital registrations. To estimate the non-fatal burden, we used data presented in primary studies, hospital discharges, and claims data from the United States, and used DisMod-MR 2.1, a Bayesian meta-regression tool, to ensure consistency between measures. Mortality, prevalence, years of life lost (YLLs) due to premature death, years lived with disability (YLDs), and disability-adjusted life-years (DALYs) were estimated. All of the estimates were reported as numbers and rates per 100,000, with 95% uncertainty intervals (UI).
Results: Globally, there were 6.8 million (95% UI 6.4–7.3) cases of IBD in 2017 compared with 3.7 million (3.5–3.9) in 1990. The age-standardised prevalence increased from 79.5 per 100,000 (75.9–83.5) in 1990 to 84.3 per 100,000 (79.2–89.9) in 2017.
The total number of deaths increased from 23,126 (95% UI 10,184–27,466) in 1990 to 38,628 (31,595–41,157) in 2017, while the age-standardised death rate (ASDR) decreased from 0.61 per 100,000 (0.55–0.69) in 1990 to 0.51 per 100,000 (0.42–0.54) in 2017. IBD-related deaths comprised 0.07% (0.06–0.07) of total all-cause deaths in 2017. IBD was the fourth leading cause of YLDs among digestive diseases in 2017.
The highest age-standardised prevalence rates in 2017 occurred in high-income North America (422.0 per 100,000 [95% UI 398.7–446.1]). The lowest prevalence rates in 2017 were observed in the Caribbean (6.73 [6.3–7.2] per 100,000). Locations with high socio-demographic index (SDI) had the highest age-standardised prevalence rates, while low SDI regions had the lowest rates, with values that were much higher and lower than global prevalence rates, respectively.
At the national level, USA had the highest prevalence rate (464.4 per 100,000 [95% UI 438–490]), followed by the UK (449.6 per 100,000 [420–481]). Vanuatu had the highest ASDR in 2017 (1.8 per 100,000 [0.81–3.2]) and Singapore had the lowest (0.08 per 100,000 [0.06–0.1]).
Conclusion: The prevalence of IBD increased substantially in many regions from 1990 to 2017, which may impose a substantial social and economic burden in the coming years. Our findings can be useful for policy makers developing strategies to tackle this disease.
Disclosure: Nothing to disclose.
LB23 GLOBAL, REGIONAL, AND NATIONAL BURDEN OF OESOPHAGEAL CANCER AND ITS ATTRIBUTABLE RISK FACTORS, 1990 TO 2017: A SYSTEMATIC ANALYSIS FOR THE GLOBAL BURDEN OF DISEASE STUDY 2017
F. Kamangar1, D. Nasrollahzadeh2,3, S. Safiri4,5, C. Fitzmaurice6,7, M. Naghavi7, R. Malekzadeh2,8
1Morgan State University, Baltimore, United States
2Digestive Oncology Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran, Iran (Islamic Republic of)
3Section of Genetics, International Agency for Research on Cancer, World Health Organization, Lyon, France
4Department of Community Medicine, School of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran (Islamic Republic of)
5Aging Research Institute, Tabriz University of Medical Sciences, Tabriz, Iran (Islamic Republic of)
6Division of Hematology, Department of Medicine, University of Washington, Seattle, United States
7Institute for Health Metrics and Evaluation, University of Washington, Seattle, United States
8Digestive Disease Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran, Iran (Islamic Republic of)
Contact E-Mail Address:malek@tums.ac.ir
Introduction: Oesophageal cancer (OC) is a common and often fatal cancer that has two main histological subtypes: oesophageal squamous cell carcinoma (OSCC) and oesophageal adenocarcinoma (OA). Updated statistics on the incidence, mortality, and disability-adjusted life-years (DALYs) caused by OC can assist policy makers in allocating resources for OC prevention, treatment, and care. This study presents the latest estimates of these statistics for 195 countries and territories between 1990 and 2017, by age, sex, and socio-demographic index (SDI).
Aims & Methods: Vital registration (nsite-years = 19,323), vital registration-sample (nsite-years = 793), verbal autopsy (nsite-years = 419), and cancer registry (nsite-years = 5497) data points combined with relevant modelling were used to estimate mortality, incidence, and burden of OC and their trends from 1990 to 2017 in Global Burden of Disease data (GBD). Mortality-to-incidence ratios (MIRs) were estimated and fed into a Cause of Death Ensemble model (CODEm) including risk factors. MIRs were used for mortality and non-fatal modelling. Estimates of DALYs attributable to the main risk factors of OC available in the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD) were also calculated. Proportion of OSCC to OC was extracted using publicly available data, and its variation was examined against SDI and available risk factors in GBD that are specific for OSCC (unimproved water source, indoor air pollution) and for OA (gastro-oesophageal reflux disease).
Results: There were 472,525 (95% UI 459,485–485,294) new cases of OC and 435,959 (424,994–447,580) deaths due to OC in 2017. Age-standardised incidence and mortality rates were 5.9 (5.7–6.1) and 5.5 (5.3–5.6) per 100,000, respectively. There were also 9.8 million (9.5–10.0) DALYs due to OC, with an age-standardised rate of 119.9 (116.9–123.0) per 100,000. Between 1990 and 2017, age-standardised incidence, mortality, and DALY rates decreased by 22%, 29%, and 33%, respectively, while, due to population growth and ageing, the total number of new cases, deaths, and total DALYs increased by 52%, 40%, and 27%, respectively. The highest incidence, mortality, and DALY rates were observed in some countries in eastern and southern sub-Saharan Africa (e.g. Malawi, Eswatini, Lesotho, and Zimbabwe), and east and central Asia (e.g. China, Mongolia, and Afghanistan). The age-standardised incidence (2.7-fold), mortality (2.9-fold), and DALY (3.0-fold) rates in 2017 were higher for men than women. Rates for both sexes were highest in age groups 50 to 90. As SDI increased, incidence and mortality rates of OC decreased. Smoking tobacco, drinking alcohol, high body mass index, low intake of fruits, and chewing tobacco were responsible for 38.9% (35.4–42.1), 33.6% (27.1–39.6), 19.4% (6.3–35.9), 19.1% (4.2–34.6), and 7.5% (5.2–9.6) of DALYs, respectively. Among these risk factors, high body mass index is an established risk factor for OA but has no proven association with OSCC risk. Countries with high OSCC:OC proportion had lower SDI, Healthcare Access and Quality Index, and indoor air pollution.
Conclusion: Despite reductions in age-standardised incidence and mortality rates, OC remains a major cause of cancer mortality and burden across the world. Oesophageal cancer has a high mortality-to-incidence ratio (0.91), requiring increased primary prevention efforts and perhaps screening in some very high-risk areas. There is substantial variation in rates across regions and countries, as well as within countries, for reasons that are unclear.
Disclosure: Nothing to disclose.
LB24 THE GLOBAL, REGIONAL, AND NATIONAL BURDEN OF STOMACH CANCER IN 195 COUNTRIES, 1990–2017: A SYSTEMATIC ANALYSIS FOR THE GLOBAL BURDEN OF DISEASE STUDY 2017
A. Etemadi1,2, S. Safiri3,4, S.G. Sepanlou5, C. Fitzmaurice6,7, M. Naghavi7, R. Malekzadeh1,5
1Digestive Oncology Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran, Iran (Islamic Republic of)
2Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, United States
3Department of Community Medicine, School of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran (Islamic Republic of)
4Aging Research Institute, Tabriz University of Medical Sciences, Tabriz, Iran (Islamic Republic of)
5Digestive Disease Research Center, Digestive Disease Research Institute, Shariati Hospital, Tehran University of Medical Sciences, Tehran, Iran (Islamic Republic of)
6Division of Hematology, Department of Medicine, University of Washington, Seattle, United States
7Institute for Health Metrics and Evaluation, University of Washington, Seattle, United States
Introduction: Stomach cancer is a major health problem in many countries. Due to geographical variations, understanding the current burden of stomach cancer and the differential trends across various locations is essential for formulating effective preventive strategies.
Aims & Methods: We used estimates from the Global Burden of Diseases, Injuries, and Risk Factors (GBD) Study 2017 (GBD 2017) to analyse the incidence, mortality, and disability-adjusted life-years (DALYs) due to stomach cancer in 195 countries and territories from 21 regions and study the changes in these estimates between 1990 and 2017. The rates were standardised to the GBD world population and reported per 100,000 as age-standardised incidence rates (ASIR), age-standardised death rates (ASDR), and age-standardised DALY rates. All estimates were generated with 95% uncertainty intervals (UIs).
Results: In 2017, more than 1.22 million (95% UI 1.19–1.25) incident cases of stomach cancer occurred worldwide and nearly 865,000 people (848,000–885,000) died of stomach cancer, contributing to 19.1 million (18.7–19.6) DALYs. The highest ASIR in 2017 were seen in high-income Asia-Pacific countries (ASIR: 29.5 [28.2–31.0]) and East Asia (ASIR: 28.6 [27.3–30.0]), with nearly half of the global incident cases living in China. Compared with 1990, in 2017 there were over 356,000 more incident cases of stomach cancer, leading to nearly 96,000 more deaths. Despite the increase in absolute numbers, the worldwide age-standardised rates of stomach cancer (incidence, death, and DALYs) have declined since 1990. The drop in the disease burden showed a correlation with improved socio-demographic index. Globally, 38.2% of the DALYs were attributable to high-sodium diet in both sexes, and in men, 24.5% of the DALYs were attributable to smoking.
Conclusion: Our findings provide insight into the changing burden of stomach cancer, which is useful in planning local strategies and monitoring their progress. To this end, specific local strategies should be tailored to each country’s risk factor profile. Beyond the current decline in age-standardised incidence and death rates, a decrease in the absolute number of cases and deaths will be possible if the burden in East Asia, where currently almost half of the incident cases and deaths occur, is further reduced.
Disclosure: Nothing to disclose.
LB25 THE GLOBAL, REGIONAL, AND NATIONAL BURDEN OF PANCREATIC CANCER AND ITS ATTRIBUTABLE RISK FACTORS IN 195 COUNTRIES AND TERRITORIES, 1990–2017: A SYSTEMATIC ANALYSIS OF THE GLOBAL BURDEN OF DISEASE STUDY 2017
A. Pourshams1, S.G. Sepanlou1,2, C. Fitzmaurice3,4, M. Naghavi4, R. Malekzadeh1,2
1Digestive Oncology Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran, Iran (Islamic Republic of)
2Digestive Disease Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran, Iran (Islamic Republic of)
3Division of Hematology, Department of Medicine, University of Washington, Seattle, United States
4Institute for Health Metrics and Evaluation, University of Washington, Seattle, United States
Contact E-Mail Address:malek@tums.ac.ir
Introduction: Worldwide, both the incidence and death rates of pancreatic cancer (PC) are increasing. Evaluation of PC burden and its global, regional, and national patterns is crucial to policy making and better resource allocation for controlling PC risk factors, developing early detection methods, and providing faster and more effective treatments.
Aims & Methods: Vital registration, vital registration-sample, and cancer registry data were used to generate the relevant estimates. Mortality, incidence, and disability-adjusted life-years (DALYs) were estimated and reported. All of the estimates were reported as counts and age-standardised rates per 100,000. 95% uncertainty intervals (UIs) were reported for all estimates.
Results: In 2017, there were 447,000 (95% UI 438–456) incident cases of PC globally. The age-standardised incidence rate was 5.0 (4.9–5.1) per 100,000 in 1990 and increased to 5.7 (5.6–5.8) per 100,000 in 2017. There was a 2.3-times increase in number of deaths for both sexes from 196,000 (193–200) in 1990 to 441,000 (433–449) in 2017. In both 1990 and 2017, 52% of total incident cases occurred in males. There was a two-times increase in DALYs due to PC, increasing from 4.4 million (4.3–4.4) in 1990 to 9.1 million (8.9–9.3) in 2017.
The age-standardised death rate of PC was highest in the high-income super-region across all years from 1990 to 2017.
In both 1990 and 2017, the highest age-standardised death rates were observed in Greenland (17.4 [95% UI 15.8–18.9] per 100,000 in 2017) and Uruguay (12.1 [10.9–13.5] per 100,000 in 2017), with substantial distance to the third-ranked country, the Czech Republic. Bangladesh (1.9 [1.5–2.3] per 100,000) had the lowest rate in 2017, while Yemen (1.3 [1.0–2.1] per 100,000) had the lowest rate in 1990.
The number of both incident cases and deaths peaked in the 65–69-year age group for males and 75–79-year age group for females. PC deaths worldwide were primarily attributable to smoking (21.2% [95% UI 18.9–23.8]), high fasting plasma glucose (8.8% [2.1–19.2]), and high body mass index (6.2% [2.5–11.5]) in 2017.
Conclusion: Globally, the burden of PC has more than doubled from 1990 to 2017. The increase in incidence of PC may continue as the population ages. Prevention strategies should focus on modifiable risk factors. Development of screening programmes for early detection and more effective treatment strategies for PC are mandatory.
Disclosure: Nothing to disclose.
LB26 THE GLOBAL, REGIONAL, AND NATIONAL BURDEN OF COLORECTAL CANCER AND ITS ATTRIBUTABLE RISK FACTORS IN 195 COUNTRIES AND TERRITORIES, 1990–2017: A SYSTEMATIC ANALYSIS FOR THE GLOBAL BURDEN OF DISEASE STUDY 2017
S. Safiri1,2, S.G. Sepanlou3, A. Delavari3, C. Fitzmaurice4,5, M. Naghavi5, R. Malekzadeh3
1Department of Community Medicine, School of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran (Islamic Republic of)
2Aging Research Institute, Tabriz University of Medical Sciences, Tabriz, Iran (Islamic Republic of)
3Digestive Diseases Research Center, Digestive Diseases Research Institute, Shariati Hospital, Tehran University of Medical Sciences, Tehran, Iran (Islamic Republic of)
4Division of Hematology, Department of Medicine, University of Washington, Seattle, United States
5Institute for Health Metrics and Evaluation, University of Washington, Seattle, United States
Contact E-Mail Address:malek@tums.ac.ir
Introduction: Data on the global, regional, and country-specific variations in the levels and trends of colorectal cancer (CRC) are required in order to understand the impact of this disease and the trends in its burden to help policy makers allocate resources.
Aims & Methods: The present study provides a status report on the incidence, mortality, and disability caused by CRC in 195 countries and territories between 1990 and 2017. Vital registration, vital registration-sample, verbal autopsy, and cancer registry data were used to generate the relevant estimates. Mortality, incidence, and disability-adjusted life-years (DALYs) were estimated and reported. All of the estimates were reported as counts and age-standardised rates per 100,000 person-years. 95% uncertainty intervals (UIs) were reported for all estimates.
Results: In 2017, there were 1.8 million (95% UI 1.8–1.9) incident cases of CRC globally, with an age-standardised incidence rate of 23.2 (22.7–23.7) per 100,000 that increased by 9.5% (4.5–13.5) between 1990 and 2017. Globally, CRC accounted for 896,000 (876.3–915.7) deaths in 2017, with an age-standardised death rate of 11.5 (11.3–11.8) per 100,000, which decreased by 13.5% (10.0–18.4) since 1990. CRC was also responsible for 18.9 million (18.4–19.4) DALYs in 2017 globally, with an age-standardised rate of 235.7 (229.7–242.0) DALYs per 100,000, which decreased by 14.5% (10.3–20.4) since 1990. Slovakia, the Netherlands, and New Zealand had the highest age-standardised incidence rates in 2017. Greenland, Hungary, and Slovakia had the highest age-standardised death rates in 2017. Numbers of incident cases and deaths were higher among males than females up to ages 80–84, with the highest rates observed in the oldest age group, 95 and older, for both sexes in 2017. SDI and HAQ Index showed a non-linear association with age-standardised DALY rate. In 2017, the three largest contributors to DALYs at the global level, for both sexes, were diet low in calcium (20.5%), alcohol use (15.2%), and diet low in milk (14.3%).
Conclusion: There is substantial global variation in the burden of CRC. Although the overall CRC death rate has been decreasing at the global level, the increasing incidence rate in most countries poses a major public health challenge across the world. The results of the present study can be useful for policy makers to carry out cost-effective interventions and to reduce exposure to modifiable risk factors, particularly in countries with high incidence levels and/or increasing trends.
Disclosure: Nothing to disclose.
14:00–15:30 / B2
Paradigm shifts in IBD treatment
LB27 HIGH VERSUS STANDARD ADALIMUMAB INDUCTION DOSING REGIMENS IN PATIENTS WITH MODERATELY TO SEVERELY ACTIVE CROHN’S DISEASE: RESULTS FROM THE SERENE-CD INDUCTION STUDY
G. D’Haens1, W.J. Sandborn2, E. Loftus, Jr3, S.B. Hanauer4, S. Schreiber5, L. Peyrin-Biroulet6, R. Panaccione7, J. Panes8, J.-F. Colombel9, M. Ferrante10, E. Louis11, A. Armuzzi12, Q. Zhou13, B. Huang13, N. Kwatra13, N. Mostafa13, T. Doan13, J. Petersson13, A. Song13, A. Robinson13, S. Danese14
1Amsterdam University Medical Centers, Amsterdam, Netherlands
2University of California San Diego, San Diego, United States
3Mayo Clinic, Rochester, United States
4Feinberg School of Medicine, Northwestern University, Chicago, United States
6Nancy University Hospital Inserm U954 Dept. of Hepato-Gastroenterology, Department of Gastroenterology, Vandoeuvre les Nancy, France
7University of Calgary, Medicine, Calgary, Canada
8Hospital Clínic Barcelona Dept. of Gastroenterology – Dept. of Gastroenterology, Hospital Clínic Bar, Dept. of Gastroenterology, Barcelona, Spain
9Icahn School of Medicine at Mount Sinai, Gastroenterology, USA, United States
10University Hospitals Leuven, Department of Gastroenterology and Hepatology, Leuven, Belgium
11University Hospital of Liège, Dept. de Gastroenterologie, Liège, Belgium
12Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy
13AbbVie Inc, North Chicago, United States
14Humanitas Research Hospital, Gastrointestinal Immunopathology, Rozzano, Italy
Contact E-Mail Address:g.dhaens@amsterdamumc.nl
Introduction: Adalimumab (ADA) is effective and well tolerated in inducing and maintaining clinical remission in patients (pts) with Crohn’s disease (CD).1 We report results from the 12-wk induction period of SERENE-CD (NCT02065570), comparing two ADA induction dosing regimens (higher [HIR] and standard [SIR]).
Aims & Methods: SERENE-CD is a Phase 3, double-blind, randomized, multicenter study of higher versus standard ADA dosing regimens for induction therapy in adult pts with moderately to severely active CD. Pts with Crohn’s Disease Activity Index (CDAI) 220–450 and endoscopic inflammation per central reading were randomized 3:2, receiving ADA either as the HIR (160 mg at Wks 0, 1, 2, and 3) or the SIR (160 mg at Wk 0 and 80 mg at Wk 2) followed by 40 mg every other week at Wk 4 through Wk 12 for both regimens. Pts were stratified by baseline (BL) C-reactive protein, prior infliximab use, and CDAI. The co-primary efficacy endpoints for induction were the proportion of pts in the intent-to-treat (ITT) population achieving clinical remission (CDAI < 150) at Wk 4, and the proportion of pts achieving endoscopic response (decrease in Simplified Endoscopic Score for Crohn’s Disease [SES-CD] > 50% from BL [or for a BL SES-CD score of 4, ≥2-point reduction from BL]) at Wk 12. Non-responder imputation was used for missing values. ADA trough serum concentrations were measured at Wks 2, 4, 8, and 12. Exposure–response relationships (ERRs) for efficacy endpoints were assessed using NONMEM 7.3. Safety assessment included collection of adverse events (AEs), vital signs, and laboratory data.
Results: A total of 514 pts were randomized, 308 and 206 into the HIR and the SIR, respectively; 92.9% and 93.2% completed induction. BL demographics were generally well balanced across the two treatment arms; overall, mean (standard deviation [SD]) CD duration was 7.3 (8.5) years, 17.3% had prior infliximab use, and 49.2% had BL corticosteroid use. There was no significant difference in the rate of clinical remission at Wk 4 (43.2% vs. 43.7%; p > 0.999) or endoscopic response at Wk 12 (42.5% vs. 39.3%; p = 0.509) between HIR and SIR, respectively. Results for ranked secondary efficacy endpoints are displayed in the table; only the proportion of pts achieving clinical remission or clinical response at Wk 12 was higher for the HIR versus the SIR at nominal p-value < 0.05. ADA serum concentrations were higher in the HIR versus the SIR (mean [SD] = 40.1 [15.0] and 11.8 [5.3] µg/mL at Wk 4 and 14.4 [7.0] and 8.2 [4.8] µg/mL at Wk 12, respectively). A clear ERR was observed for endoscopic response at Wk 12 where higher ADA concentrations resulted in higher response, but not for clinical remission at Wk 4. The observed safety profile was similar between the HIR and SIR groups, including AEs of special interest. No new safety signals or unexpected trends were identified.
Conclusion: The results from SERENE-CD show no additional benefit of ADA HIR versus SIR in inducing clinical remission at Wk 4 or endoscopic response at Wk 12. A clear ERR was observed for endoscopic response. Both dosing regimens were generally safe and well tolerated. The maintenance part of the study is ongoing.
Disclosure: Geert D’Haens: Consulting and/or lecture fees from AbbVie, ActoGeniX, AIM, Allergan, Amgen, Arena Pharmaceuticals, Boehringer Ingelheim GmbH, Celgene/Receptos, Celltrion, Cosmo; and others. William J. Sandborn: Research grants from AbbVie, Amgen, Atlantic Healthcare Limited, Celgene/Receptos Genentech, Gilead Sciences, Janssen, Lilly, and Takeda; consulting fees from AbbVie, Allergan, Amgen; and others. Edward V. Loftus Jr: Consultancy: AbbVie, Allergan, Amgen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Celltrion Healthcare, Eli Lilly, Genentech, Gilead, Janssen, Pfizer, Takeda, and UCB; research support: AbbVie, Amgen, Genentech, Gilead; and others. Stephen Hanauer: Consulting fees from AbbVie, Actavis, Allergan, Amgen, Arena, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Celltrion, Ferring, Genentech, Gilead, GlaxoSmithKline, Hospira, Janssen; and others. Stefan Schreiber: Consultancy: AbbVie, Falk Pharma, Ferring, Genentech, GlaxoSmithKline, MSD, Pfizer, Shire, and Takeda. Laurent Peyrin-Biroulet: Lecture fee(s): AbbVie and Merck; Consultancy: AbbVie, Amgen, Biogen, Biogaran, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Celltrion, Ferring, Forward Pharma GmbH, Genentech; and others. Remo Panaccione: Consultant and/or lecture fees from AI4GI, AbbVie, Arena Pharmaceuticals Amgen, Atlantic Healthcare, BioBalance, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Coronado Biosciences; and others. Julian Panés: Financial support for research: AbbVie and MSD; lecture fee(s): AbbVie, Ferring, Janssen, MSD, Pfizer, Shire Pharmaceuticals, Takeda, and Theravance; consultancy: AbbVie, Arena Pharmaceuticals, Boehringer Ingelheim; and others. Jean-Frederic Colombel: Consultant, advisory board member, or speaker for AbbVie, Bristol-Myers Squibb, Ferring Pharmaceuticals, Genentech, Giuliani SPA, Given Imaging, Merck, Millennium Pharmaceuticals; and others. Marc Ferrante: Research grant: Amgen, Biogen, Janssen, Pfizer, and Takeda; consultancy: AbbVie, Boehringer Ingelheim, Ferring, Janssen, Mitsubishi Tanabe, MSD, Pfizer, and Takeda; speakers fee: AbbVie, Boehringer Ingelheim; and others. Edouard Louis: Received honoraria for lectures or consultation from Abbott, AstraZeneca, Centocor, Falk, Ferring, Millennium, Schering-Plough, and UCB; research grants from AstraZeneca and Schering-Plough. Alessandro Armuzzi: Consultant or advisory member for AbbVie, Allergan, Amgen, Biogen, Bristol-Myers Squibb, Celgene, Celltrion, Ferring, Hospira, Janssen, Lilly, MSD, Mundipharma, Mylan, Pfizer, Roche; and others. Silvio Danese: Financial support for research: AbbVie, Amgen, Genentech, Gilead, Janssen, Medimmune, Pfizer, Receptos, Robarts Clinical Trials, Seres Therapeutics, Takeda, and UCB; consultancy: AbbVie, Allergan, Amgen; and others. Qian Zhou, Bidan Huang, Nisha Kwatra, Nael Mostafa, Thao Doan, Joel Petersson, Alexandra Song, and Anne Robinson: AbbVie employees, and may own AbbVie stock and/or options.
1. Sustained clinical remission at both Wks 4 and 12, n (%)
119 (38.6)
72 (35.0)
0.305
2. Clinical remission at Wk 4 and endoscopic response at Wk 12, n (%)
67 (21.8
42 (20.4)
0.675
3. Clinical remission at Wk 12, n (%)
192 (62.3)
106 (51.5)
0.008*
4. Proportion of pts taking CS at BL who discontinued CS and achieved clinical remission at Wk 12, n/N (%)
80/153 (52.3)
47/100 (47.0)
0.309
5. Endoscopic remission (SES-CD ≤ 4 and ≥2-point reduction versus BL, and no subscore >1 in any individual variable) at Wk 12, n (%)
88 (28.6)
54 (26.2)
0.694
12. Clinical response (decrease in CDAI ≥ 70 points from BL) at Wk 12, n (%)
257 (83.4)
154 (74.8)
0.015*
*p-value < 0.05; although two endpoints had nominal p-values < 0.05, the ranked secondary endpoints did not achieve statistical significance between HIR and SIR based on prespecified fixed-sequence multiple testing procedure. aComparisons based on Cochran–Mantel–Haenszel test adjusted for previous IFX use (or prior anti-TNF use for subjects randomized under original protocol), hs-CRP at BL (<10 mg/L, ≥10 mg/L), and Crohn’s disease severity at BL (CDAI ≤ 300, CDAI > 300). BL: baseline; CDAI: Crohn’s Disease Activity Index; CS: corticosteroid; HIR: higher induction dosing regimen; hs-CRP: high-sensitivity C-reactive protein; IFX: infliximab; ITT: intent-to-treat; SES-CD: Simplified Endoscopic Score for Crohn’s Disease; SIR: standard induction dosing regimen; Wk: week.
Reference
LichtensteinGR, et al.Efficacy and Safety of Adalimumab in Crohn's Disease. Ther Adv Gastroenterol2008; 1: 43–50.