
Abstract
Crohn’s disease affects those individuals with polygenic risk factors. The identified risk loci indicate that the genetic architecture of Crohn’s disease involves both innate and adaptive immunity and the response to the intestinal environment including the microbiome. Genetic risk alone, however, predicts only 25% of disease, indicating that other factors, including the intestinal environment, can shape the epigenome and also confer heritable risk to patients. Patients with Crohn’s disease can have purely inflammatory disease, penetrating disease or fibrostenosis. Analysis of the genetic risk combined with epigenetic marks of Crohn’s disease and other disease associated with organ fibrosis reveals common events are affecting the genes and pathways key to development of fibrosis. This review will focus on what is known about the mechanisms by which genetic and epigenetic risk factors determine development of fibrosis in Crohn’s disease and contrast that with other fibrotic conditions.
Introduction
Disease pathogenesis results from the heritable risk that accrues from alterations in DNA sequence, risk polymorphisms, and from alterations in the epigenome that control gene expression when exposed to environmental change. Epigenetic control of gene expression is exerted through modification of DNA regulatory elements or enhancers that induce transition of condensed heterochromatin, where gene transcription is inhibited by histone modifications and DNA methylation, to euchromatin, where genes are accessible for transcription. Gene expression is also controlled by small non-coding interfering RNAs, microRNAs, which post-transcriptionally regulate gene expression. Crohn’s disease is a polygenic disorder with more than 200 risk loci identified by genome-wide association studies (GWAS). However, understanding the risk of disease development or expression of a specific phenotype of Crohn’s disease in a patient is not predicted or understood solely by genetic risk. Examination of the epigenetic changes associated with development of fibrosis in Crohn’s disease and fibrosis in other organs, including the lungs, heart, liver, and kidneys reveals patterns that are common to all. This review will focus on what is known about the mechanisms by which genetic and epigenetic risk factors determine development of fibrosis in Crohn’s disease and contrast that with other fibrotic conditions.
Genetics
Inflammatory bowel diseases, Crohn’s disease, and ulcerative colitis are polygenic diseases for which ∼200 risk loci have been identified.1,2 The mostly highly significant genetic associations are with the intracellular bacterial sensor NOD2, defective autophagic responses with ATG16L1 and IRGM, and with the IL-23R, indicating the genetic architecture of Crohn’s disease involves both defective innate and adaptive immune responses to intestinal microbiota. 1 To date, a deeper analysis of GWAS data has not fully revealed a genomic basis that accounts for individual Crohn’s disease phenotypes.3,4 An approach using multi-locus genetic risk scores has improved the genetic risk assessment of inflammatory bowel disease (IBD) but also indicates that rather than established risk variants, other independent variables modulate disease.5,6 Ethnic variations in the complement of associated risk loci does not account for ethnic variations in disease location or behavior in Crohn’s disease.2,7 Purely genetic models of Crohn’s disease are prone to underestimate the interactions among risk loci, termed epistasis. 8 Epistatic components need to be integrated into these models by estimating the contribution of non-genetic factors, termed missing heritability, which can be accounted for by epigenetics.9,10
Examination of genetic risk loci by pathway analysis or gene ontogeny identifies groups of polymorphisms likely to play a role in pathogenesis of fibrostenosis. TGF-β is a key cytokine that is central to the development of fibrosis. The TGF-β pathway includes identified risk variants in Smad3 and Smad7, variants in the Janus-activated kinase (Jak)-Tyk2-STAT3 pathway that regulates TGF-β expression in these cells and the negative feedback of this pathway, SOCS3.11,12 The events lead to development of fibrosis in mesenchymal cells: fibroblasts, myofibroblasts and smooth muscle, the cell types that, once activated in Crohn’s disease, produce autocrine TGF-β1 and are responsible for extracellular matrix production. 13 The functional outcomes of mutations in these key GWAS risk loci that mechanistically result in TGF-β1-dependent fibrosis are distinct from the outcomes of mutations leading to initial and sustained inflammation in epithelial and immune cells in the intestine. In the case of TGF-β signaling, Smad7 is increased in epithelial and immune cells, inhibiting Treg responses, whereas Smad7 is diminished in subepithelial myofibroblasts and allows sustained TGF-β signaling and extracellular matrix production.14–16
Other risk loci have been identified that confer risk of fibrostenotic disease that involve other pathways leading to fibrosis in the intestine. The 5T5T polymorphism at the matrix metalloprotein-3 (MMP3) gene increased the risk of developing fibrostenotic complications. 17 The MMPs and tissue metalloproteinases (TIMPs) are key regulators of the balance between extracellular matrix deposition and degradation. Homozygosity for the rs1363670 G-allele near IL-12B is an independent risk factor for development of fibrostenosis, and for a shorter time to critical stricture formation in the ileum. 18 Other risk alleles have been identified in patients with penetrating disease. The Montreal classification is hierarchical, whereby patients may present with penetrating disease that is the result of underlying fibrostenosis. Identifying Montreal Class B2 fibrostenotic Crohn’s disease, however, as distinct from patients with Montreal Class B1 inflammatory and Montreal Class B3 penetrating Crohn’s disease, is difficult but of crucial importance in understanding risk loci and susceptibility of a particular phenotype. 19
Epigenetics
The identified genetic factors and susceptibility loci account for only 13.6% of disease variability and no more than 25% of the genetic risk in Crohn’s disease.1,2 Epigenetic processes translate environmental events associated with genetic risk into regulation of chromatin, which shapes the expression of genes, and thereby the activity of specific cell types that participate in disease pathophysiology. Epigenetic mechanisms are emerging as key mediators of the effects of both genetics and the environment on gene expression and disease. 20 In addition to a set of inherited epigenetic marks, there are likely non-heritable epigenetic marks that are more dynamic and change in response to environmental stimuli. 21 In Crohn’s disease interaction of the environment, including the intestinal microbiome and metabolome, with the susceptible patient’s genome and immune system shape the epigenome. These non-genetic effects that alter gene expression and function are implied by the results of multi-locus genetic risk analyses and represent the missing heritability in GWAS.5,6
Epigenetics is defined as a “stably inherited phenotype that results from mechanisms other than changes in DNA sequence”. 11 Although initially an individual’s epigenome was not thought to be heritable, there is now increasing evidence that epigenetic inheritance can persist for multiple generations. 22 Evidence from a number of lines of investigation demonstrates epigenetic heritability from cell to cell during mitosis, from generation to generation during meiosis, and includes true transgenerational inheritance, 23 which means transmittance of information from one generation to the next that affects the traits of offspring without alteration of the sequence of DNA. Such mechanisms have been shown to include incomplete erasure of DNA methylation, parental effects, transmission of distinct RNA types (e.g. mRNA, non-coding RNA, miRNA), and persistence of subsets of histone marks. 23 Epimutations, that is, epigenetic changes that are sustained in the germ line, can be transmitted in a true intergenerational fashion by surviving the developmental reprogramming that erases epigenomic changes present in the parent. This mechanism has been shown to be operative in animal models of liver fibrosis. Remodeling of DNA methylation and histone acetylation in offspring of mice harboring epigenetic changes altering TGF-β1 expression that results in liver fibrosis is lowered in male F1 and F2 generations through a process termed suppressive adaptation. 24 Humans with milder non-alcoholic fatty liver disease have hypomethylation of the anti-fibrogenic factor PPAR-γ promoter compared with patients with more severe fibrosis, lending support to this notion. All these aforementioned findings suggest transmission of an epigenetic suppressive adaptation that can help offspring better adapt to future hepatic insults that might result in fibrosis. Suppressive adaptation, however, was not seen in the setting of renal fibrosis. 24
Even though all cells within the intestine or an organism share a common genome, gene expression in an individual cell type is regulated by the unique epigenetic events that affect that cell type, and may be distinct from neighboring cell types. This can account for the sometimes contradictory epigenetic mechanisms that are identified as regulating gene expression in different cell types such as epithelial, immune, and mesenchymal cells. Thus understanding the mechanisms regulating gene expression in a cell type critical to a disease process, for example mesenchymal cells and fibrosis, based on an epigenetic analysis of DNA obtained from heterogeneous cell populations can be difficult.
Genes that can be regulated by epigenetic changes in the development of organ fibrosis.
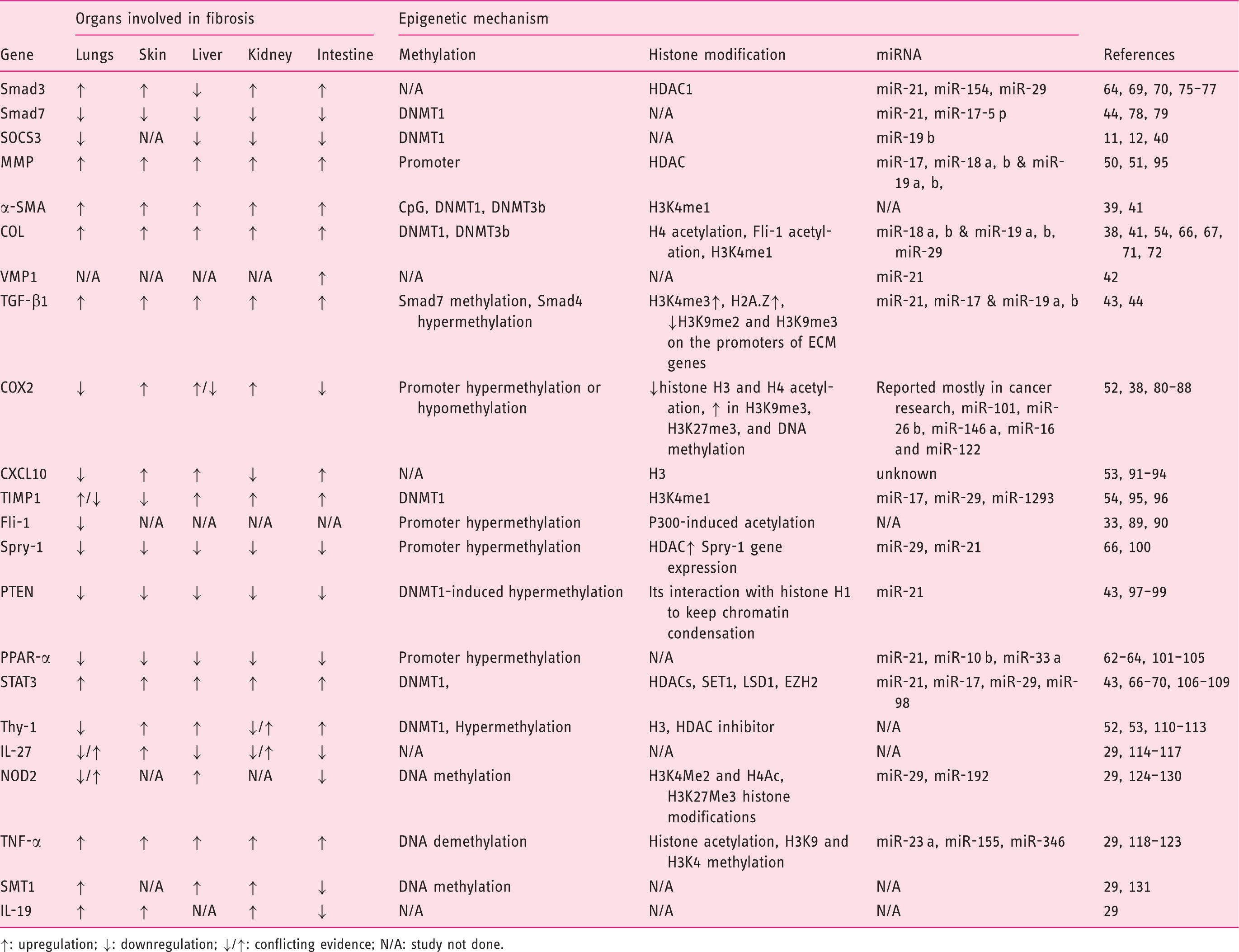
↑: upregulation; ↓: downregulation; ↓/↑: conflicting evidence; N/A: study not done.
DNA methylation
Methylation of cytosine by replacement of the hydrogen in position 5 (5MeC) in the context of CpG dinucleotides that are clustered in CpG islands is a common DNA modification. Of the 28 CpG dinucleotides present in the human genome, 60–80% are methylated. 25 Methylation typically, but not always, represses gene expression by either interfering with the binding of transcription factors to their DNA binding sites or recruiting methyl-CpG-binding proteins that attract histone and chromatin-modifying enzymes. DNA methyltransferases (DNMT)-1 and DNMT-3a and 3b are the primary enzymes responsible for methylation of CpG islands. 26 DNMT-1 is a maintenance methyltransferase, whereas DNMT-3a and 3b are de novo methyltransferases. Methylation is reversed by two processes, active and passive demethylation. The ten-eleven translocation methylcytosine dioxygenase (TET) family of enzymes function to catalyze active demethylation via 5MeC hydroxymethylation (5HMeC) which attracts DNA excision and repair machinery, restoring DNA to a demethylated status. 27 Passive demethylation occurs when maintenance methylation is absent and progressive dilution of 5MeC occurs during DNA replication. 28
DNA methylation and fibrosis
Alterations of DNA methylation have been examined in a number of disease processes that result in tissue fibrosis including systemic sclerosis, pulmonary and cardiac fibrosis, hepatic fibrosis, and intestinal fibrosis in Crohn’s disease.21,29–35 Hypermethylation of specific genes as well as global changes in DNA methylation have been identified in these organ systems. Two genomic studies in patients with idiopathic pulmonary fibrosis (IPF) demonstrated extensive DNA methylation changes in the control of IPF gene expression.36–38 Different levels of CpG island methylation are present in specific genes regulating a fibroproliferative phenotype in IPF, and myeloproliferative diseases, via miR-17∼92, involve an increased DNMT-1-mediated feedback loop involving both microRNAs and DNA methylation.39,40 Notably altered CpG island methylation in the α-smooth muscle actin (α-SMA) promoter was present in pulmonary fibroblasts and myofibroblasts in patients with IPF. 41 A core set of genes known to be related to fibrosis, including several collagens, were differentially methylated in patients with progressive renal fibrosis compared with controls. 42 Recently, in a rat model of hypoxia-induced cardiac fibrosis, global hypermethylation of gene expression was observed along with upregulation of both DNMT-1 and DMNT-3b that was associated with upregulation of collagen and α-SMA in renal fibroblasts. 43
Genome-wide methylation profiling in patients with IBD has identified numerous sites that are differentially methylated between cases and controls. 30 The most highly statistically significant include genes controlling altered immune activation, responses to luminal bacteria, and regulation of the Th17 pathway. 29 A significant enrichment in DNA methylation was seen within 50 kb of several Crohn’s disease GWAS risk loci including IL-27, IL-19, tumor necrosis factor (TNF), Soluble latent membrane-type 1 (SMT1) and NOD2. In this study by Nimmo and colleagues, methylation status was predictive of disease activity. 29 In pediatric Crohn’s disease, Adams et al. provided evidence that four of the most differentially methylated regions resided in proximity to the vacuole membrane protein-1 (VMP1) GWAS locus. 44 VMP1 is a putative transmembrane protein that has been reported to be involved in different biological events including autophagy, cell adhesion, and membrane translocation. 45 The microRNA (miR)-21 gene lies within the VMP1 gene. They share a common transcription start site and promoter region, but pri-miR-21 possesses its own unique promoter, thus VMP-1 and pri-miR-21 can be differentially transcribed. Primary miRNA (pri-miRNA) with about 100 nucleotides is transcribed from miRNA genes in the nucleus by RNA polymerase II and further processed into pre-miRNA by a microprocessor complex. Our own recent work has demonstrated that the increased transcription of miR-21 in muscle cells and myofibroblasts of patients with fibrostenotic Crohn’s disease determines the sustained TGF-β1 signaling that results in excess collagen and extracellular matrix production and fibrosis. 45 This process uniquely characterizes patients with Montreal Class B2 fibrostenotic Crohn’s disease as distinct from patients with Montreal Class B1 inflammatory and Montreal Class B3 penetrating Crohn’s disease.19,45,46
Histone modifications of DNA and post-translational modification of proteins
Histones are also key players in epigenetics. The four core histones, H2a, H2B, H3, and H4 associate as two H2A–H2B dimers and a H3–H4 tetramer that comprise the nucleosome. 47 Adjacent nucleosome octamers are separated by ∼50 kb of DNA with the linker histone, H1 interposed between. All histones are also subject to post-translational modifications on their tail regions including phosphorylation, acetylation, methylation, ubiquitination, SUMOlyation, and ADPribosylation. These post-transcriptional modifications of histone contribute to the transcriptional state of the genomic DNA. Generally euchromatin, open or lightly packed chromatin with accessible DNA and actively transcribed genes, and heterochromatin, condensed or tightly packed inaccessible chromatin, are each characterized by different levels of specific histone acetylation and/or methylation and position along the genome in promoter regions or intron/exon regions.48,49 Histone-modifying enzymes catalyze the post-translational modification of histones and non-histone proteins. This large group of enzymes includes histone acetyltransferases (e.g. p300/CBP) and histone deacetylases (e.g. HDACs), and lysine methyltransferases (e.g. LSD). 50 The transcription of a gene, therefore, is regulated by the cumulative influence of multiple histone modifications that results from the activity of histone-modifying enzymes. Data from the ENCODE project has identified key histones and their modifications that have become the most highly studied for their ability to control accessibility of chromatin and thereby regulation of gene expression (Table 1). 51
Histone modification and fibrosis
Both histone acetylation and deacetylation are linked to the development of pulmonary fibrosis. It is worth noting that H3 hyperacetylation through decreased expression of histone deactylase is consistently associated with pulmonary fibrosis.52,53 This regulation of HDAC expression in the lungs results in TGF-β-induced myofibroblast differentiation, and excess collagen and matrix metalloproteinase-1 production. 53 Acetylation levels of H3 also regulate the expression of cyclooxygenase-2, IFN-gamma-inducible protein 10 (CXCL10), and Thy-1 cell surface antigen, all of which are integral to the development fibrosis in the lung.54,55 A similar process in hepatic stellate cells regulates expression of profibrotic genes including α-SMA, collagen I, tissue inhibitor of metalloproteinases1, and TGF-β1 via Histone3 lysine4 methyltransferase I. 56 In systemic sclerosis, increased p300 acetyl transferase activity induces acetylation of Fli-1 proto-oncogene, thereby relieving the transcriptional repression of collagens IαI and Iα2, the major collagen species in fibrosis. 33
Differential patterns of histone H3 and H4 acetylation have been identified in Crohn’s disease.57,58 Mokry et al. recently provided evidence that many of the GWAS risk loci overlap with DNA regulatory elements in the intestine, particularly histone H3 lysine 27 (H3K27ac) but also p300, which is responsible for H3K27 acetylation and H3K4me1. 59 Sadler et al. have demonstrated that collagen Iα2 expression induced by the cytokines interleukin-1β, TNF-α, and TGF-β is regulated by hyperacetylation of histone H4. 60
Small or non-coding RNA interference
RNA interference of gene expression by microRNA (miR), small ∼18–24 nucleotide non-coding single-stranded RNA molecules, is implicated in the epigenetic regulation of fibrosis.61,62 In general, miRs post-transcriptionally repress gene expression by targeting mRNA for degradation. miR genes are located throughout the genome. They can be found in introns of coding regions, in introns or exons of non-coding genes, or in intergenic regions. In some cases they are transcribed independently from their own specific promoters, as is the case with primary microRNA-21 (pri-miR-21). 63
MicroRNA and fibrosis
A number of miRs have been identified that have a similar role in the regulation of fibrosis in the lung, liver, heart, kidney, or skin in addition to the intestine. While these miRs can have organ and tissue-specific regulation and effects, two are consistently associated with fibrosis and with the expression of TGF-β, miR-21, and miR-29. miR-21 is pro-fibrotic and is implicated in the transcriptional regulation of Sprouty homolog 1 (Spry-1), phosphatase and tensin homolog (PTEN), peroxisome proliferator-activated receptor-α (PPAR-α), signal transducer and activator of transcription-3 (STAT3), and Smad7.45,64–66 It is worth noting that miR expression can itself be subject to epigenetic regulation. Transcription of miR-21, as noted above for example, is regulated by the methylation level of its promoter. 67 miR-29 a,b,c are anti-fibrotic and are implicated in the suppression of collagen expression, MMP, and Spry1 expression.45,68–72 miR-29 expression is down-regulated by the TGF-β-dependent Smad3 transcription factor. The miR17∼92 cluster is also an important determinant of fibrosis. Transcribed from this cluster are miRs that can target key proteins in fibrosis including collagen IαI (miR-18 a,b and miR19a,b), TGF-β (miR-17 and miR-19 a,b), and MMPs (miR-17, miR-18 a,b and miR-19 a,b).39,73,74
Summary
GWAS analysis of Crohn’s disease has identified numerous risk loci that account for up to 25% of the genetic risk. Recent investigation of the epigenome indicates differential changes in DNA methylation patterns, histone modifications, and differential expression of miRs can further contribute to the ‘heritable’ risk of developing fibrostenotic Crohn’s disease. Integration of genetic susceptibility with changes in the epigenome associated with the development of organ fibrosis may lead to a greater understanding of the heritable risk of Crohn’s disease and open the door to target therapeutically critical processes that prevent or reverse the development of fibrosis.
For progress to be made in Crohn’s disease, efforts to understand the epigenome and the changes that relate to the identified risk loci and their associated pathways, and thus the missing heritability of fibrosis, will be needed. This understanding will only come from exploration of strictly phenotyped and genotyped patients and in individual cell types relevant to fibrosis.
Footnotes
Conflict of interest
None declared.
Funding
This work was supported by DK49691 from NIH: National Institutes for Diabetes, Digestive and Kidney Diseases (JFK).