
Abstract
Congenital hiatal hernia is a rare congenital defect and often occurs at a sporadic basis, but familial cases have also been reported. Here, we report on a 3-year-old male patient of Middle-Eastern descent, diagnosed at 5 months of age patient presenting with a congenital hiatal hernia, vermis hypoplasia manifested by axial hypotonia and horizontal nystagmus, preauricular tag, and dysmorphic features with negative genetic mutations, not fitting any reported association or syndrome, suggesting the potential existence of a novel disease entity and highlighting the necessity for further exploration into rare genetic conditions for comprehensive patient care and syndrome characterization.
Keywords
Introduction
Congenital hiatal hernia (CHH) is a herniation of abdominal elements through the esophageal hiatus of the diaphragm and into the mediastinum caused by a disorder in the diaphragm’s embryonic development, and they are variable in location and size.1–3 CHH is a rare congenital defect and often occurs at a sporadic basis, but familial cases have also been reported. 4 Four types have been defined in the SAGES guidelines (2013); Type I: sliding hernia where the gastroesophageal junction migrates above the diaphragm, Type II: rolling, or paraesophageal hernia where the gastroesophageal junction remains in place, but a portion of the fundus herniates through the hiatus adjacent to the esophagus, Type III: combined sliding-rolling hernia, and Type IV: hiatal hernia with presence of other structures such as omentum, colon, or small bowel within the hernial sac. 5 It is associated with a variable degree of pulmonary hypoplasia (PH) and persistent pulmonary hypertension. 6 Advancements in prenatal screening, particularly with the introduction of ultrasound, have enhanced the prenatal detection of CHH globally. 7
These hernias can be either isolated or complex. Complex hernias are diagnosed alongside additional abnormalities, accounting for around 40% of cases, with nearly 30% attributed to genetic factors. 8 Isolated cases, on the other hand, occur in 60% of cases, with less than 5% having a genetic basis. 8 Among associated abnormalities, congenital heart disease is the most common. 9 Central nervous system anomalies, found in 5%–10% of non-syndromic CHH cases, typically include neural tube defects and hydrocephalus. 10 CHH specifically often occurs on a sporadic basis, but familial cases have also been reported. The mechanism of development is not well understood, and no specific genetic factors have been implicated to date. 4
Herein, we present a rare case of CHH, specifically a sliding hiatus hernia diagnosed at 5 months of life for a male baby, presenting with dysmorphic features and cerebellar hypoplasia. Comprehensive genetic studies (microarray and whole exome) showed no abnormalities. This study seeks to raise awareness among pediatricians and surgeons regarding the diverse clinical manifestations observed in children with CHH, potentially highlighting a novel disease.
Case presentation
We report a rare presentation of CHH in a male infant of Middle-Eastern descent, diagnosed at 5 months of age. From birth, the patient exhibited mild symptoms, including moderate milk vomiting after each feeding and choking. The patient was delivered at 38 weeks of gestation via cesarean section due to complications arising from an attempted external cephalic version to consanguineous parents (first-degree cousins). At birth, he weighed 2.96 kg (21st percentile), had a head circumference of 35 cm (99th percentile), a length of 49 cm (32nd percentile), and Apgar scores of 8 and 9 at 1 and 5 min, respectively. He required no oxygen therapy or neonatal intensive care unit admission, received the hepatitis B vaccine and vitamin K, and was discharged home 24 h post-delivery. The 23-year-old mother had an uneventful antenatal history, although no detailed ultrasound was performed due to financial constraints.
At 5 months, he was hospitalized due to a persistent fever lasting 14 days. A routine fever work-up, including a chest X-ray, revealed bowel loops in the chest. A computed tomography scan confirmed a hiatal hernia containing more than two-thirds of his stomach, with mild hypoplastic changes in the lung and prominent esophageal dilation. A laparotomy with fundoplication was performed, complicated by an esophageal injury that was subsequently repaired.
Postoperative upper gastrointestinal studies revealed a stricture in the distal esophagus with marked proximal dilation. A 3-cm air-filled cavity in the region of the left lower diaphragm, which took contrast, indicated a probable fistula communicating with the distal esophagus on the left side. The patient was referred to a specialized hospital due to the failed hernia repair. An exploratory laparotomy revealed foregut adhesions, friable tissue, and recurrent stomach herniation, necessitating the placement of a gastrostomy.
The patient exhibited progressively prominent dysmorphic features. A comprehensive work-up was recommended to establish a diagnosis. At 6 months of age, his physical examination revealed a high, broad forehead, depressed nasal root, broad nasal bridge, triangular face, large bulbous nasal tip, anteverted nostrils, long philtrum with a deep groove, bilateral epicanthal folds, low-set ears, a left-sided ear pit and tag, high arched palate, retrognathia, and a large mouth with sparse scalp hair.
Neurologically, he had axial hypotonia with non-exaggerated deep tendon reflexes. Ophthalmic examination showed horizontal nystagmus. Hearing tests were normal, and no heart murmur was detected. Abdominal ultrasound showed no abnormalities, organomegaly, inverted nipples, abnormal fat distribution, or skin pigmentation. The patient also had hypospadias.
A brain magnetic resonance image at 10 months (see Figure 1) revealed vermis hypoplasia, a large cisterna magna, and a thin corpus callosum. Genetic studies, including microarray and whole exome sequencing, revealed a normal male karyotype, with no known suspected pathogenic variants detected. Despite these genetic results, consultants suspected a possibly novel genetic disorder. Chest computed tomography (CT) scan in transverse, sagittal, and coronal views from August 2021 demonstrates esophageal dilation, herniation of the stomach into the thoracic cavity, and mild pulmonary hypoplasia. These findings are indicative of structural anomalies associated with a hiatal hernia (Figure 2).

T2-weighted magnetic resonance images, in coronal and sagittal view, showing vermis hypoplasia and a thin corpus callosum.
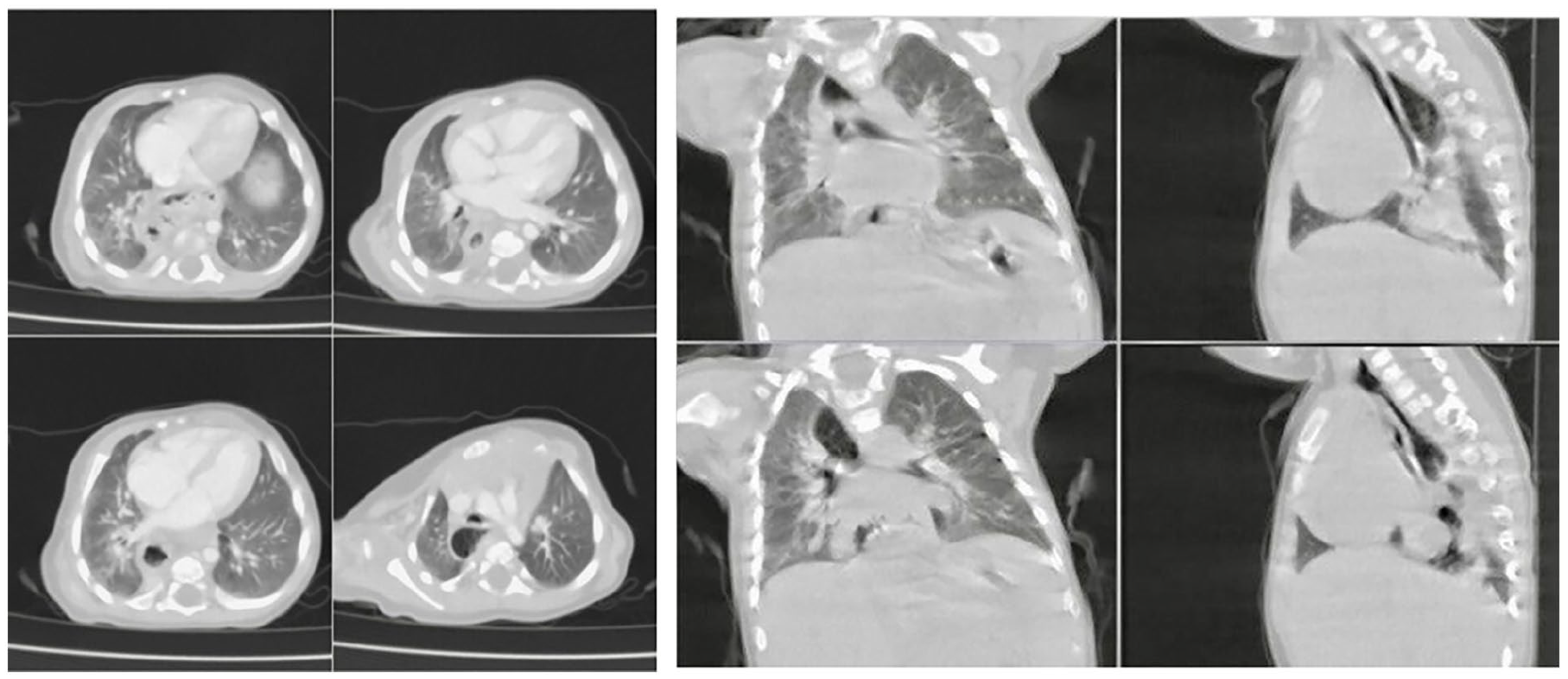
Transverse, coronal, and sagittal views show stomach herniation, significant dilatation of the esophagus, and hypoplastic changes of the right lung.
Discussion
CHH is a rare congenital defect of the diaphragm crura around the esophagus. The mechanism of development is not well understood, and no specific genetic factors have been implicated to date. Chang et al. reported the gene duplication of 1.09 Mb 9q22 and 2.73 Mb 9q22 in two families with 9q22 duplication and CHH in four individuals. 4 However, to our knowledge, neither further information on associations was present to consider, nor other possibly implicated genetic mutations.
With CHH in general, it oftentimes occurs with many genetic mutations, such as GATA4 and LRP2 and has been reported in association with syndromes such as Pallister–Killian, 8p23.1 deletion, Fryns, and Cornelia de Lange. The most frequent aneuploidies associated with it are trisomies such as 21, 18, and 13. 10
Possible syndromes were considered, such as Fryns syndrome, the criteria of which the patient did not fulfill. It is a rare syndrome characterized by diaphragmatic defects, characteristic facial appearance, short distal phalanges of the fingers and toes, PH, and associated anomalies. Survival beyond the neonatal period is rare. 11 For diagnosis, six clinical criteria of Lin et al. are required, of which is a molecular proband with suggestive findings and biallelic pathogenic variants in PIGN identified by molecular genetic testing. 12 He does not fulfill the criteria, having neither the relevant family history, characteristic facial appearance, nor short distal phalanges. And most importantly, the negative genetic profile.
The preauricular tag raised concerns for Branchio–Oto–Renal syndrome, which describes malformations of the ear associated with hearing impairment, branchial fistulae and cysts, and renal disorders. The diagnosis is established in a proband with the clinical features and/or heterozygous pathogenic variants in EYA1, SIX1, or SIX5 identified on molecular genetic testing. 13 The patient’s hearing is not impaired, and the renal system is intact, with no family history and no branchial arch anomalies.
As reviewed by Accogli et al., 14 vermis hypoplasia refers to a decreased volume of the vermis sparing the cerebellar hemispheres. Clinical features associated with isolated vermis hypoplasia and cerebellar hypoplasia (VH and CH) are variable and may include hypotonia, truncal, appendicular and gait ataxia, as well as eye movement abnormalities (saccadic pursuit, nystagmus, and oculomotor apraxia). Data evaluated by Bolduc and Limperopoulos suggested that children with VH, pontocerebellar hypoplasia type II, and cerebellar agenesis experience moderate-to-severe global developmental delays.15,16
A whole exome sequencing study in a large cohort of individuals with cerebellar malformations elucidated an underlying genetic cause in 51% of the CH cases. 17 A dysmorphological assessment is crucial for the diagnosis of an underlying syndrome displaying VH/CH, such as CHARGE syndrome, Galloway–Mowat syndrome, or MARCH syndrome associated with bi-allelic CEP55 mutations, 18 2p15p16.1 deletion, 19 Cri-du-chat syndrome, 6q27 terminal deletions, and Xq28 microduplications. 19
The patient’s negative genetic mutation results pose a challenge to our understanding of genetics and their influence on the clinical picture. It also highlights possible multimodal inheritance factors, as the patient’s parents are first-degree cousins. It prompts a new approach to viewing environmental and genetic factors influencing the patient and his genetic phenotype from conception to birth.
Our patient presents a rare and complex clinical scenario of dysmorphic features vermis hypoplasia, enlarged cisterna magna and thin corpus callosum, and CHH with esophageal dilation, despite the absence of detectable genetic mutations commonly associated with such manifestations. This clinical vignette poses a diagnostic challenge, not merely due to the objective nature of the anomalies, but as each component may be indicative of a distinct genetic or syndromic etiology or may very well suggest the possibility of a new syndrome.
Many possibilities lie in explaining this rare and complex presentation. Yet the patient fits no pre-determined image of an anomaly or a described association and remains without an established, confirmed diagnosis to guide his management throughout the years, whether preemptive screening or prophylactic management. Herein lies the importance of reporting medical cases and the ultimate goal of this case report.
The patient’s road to diagnosis has been hindered by a lack of similar cases to guide his treatment and follow-up, and the absence of a decisive pathology which would allow medical professionals to preemptively treat him and devise personalized protocols and treatment plans. Additionally, the detailed prenatal ultrasound would have alerted medical professionals to his hernia, and prevented the delay in its treatment and the resulting complications that currently affect his quality of life.
Given the rarity of the observed combination of features without a clear genetic basis, we should consider the possibility of novel or less-characterized genetic variants, including non-coding mutations or structural chromosomal abnormalities. Advanced genomic techniques such as whole-genome sequencing may unveil subtle genetic alterations that standard sequencing methods may overlook and may nominate a likely genetic culprit to consider.
The long-term prognosis and neurodevelopmental outcomes for infants with this unique combination of features remain uncertain, but inferring from each anomaly’s reported data, it is sure to be complicated by significantly higher levels of morbidity, if not mortality, in the future. As such, continued follow-up and multidisciplinary collaboration are essential for monitoring the child’s progress, identifying potential new comorbidities, and managing them before they pose significant risks to the patient’s life.
Conclusion
This case underscores the complexity of congenital anomalies, probing us to re-evaluate our understanding of genetics and their effects on clinical manifestations. One cannot underscore the need for a thorough and integrated approach to diagnosis when faced with a rare combination of symptoms and reporting on them. Further research, collaboration, and the application of advanced genomic techniques are critical to unraveling the underlying genetic and environmental factors contributing to such intricate clinical presentations.
Footnotes
Acknowledgements
The authors are grateful to Dr. Maram Bandak for revising the article and support throughout the research process.
Author contributions
The authors confirm their contribution to the article as follows: study conception and design: R.M., M.S., and A.S.; data collection: E.B., I.S., and H.A.; analysis and interpretation of results: A.S., I.S., and M.S.; draft manuscript preparation: M.S., I.S., A.S., H.A., E.B., and B.B.; reviewing and editing: M.S and A.S.; validation: R.M. All authors reviewed the results and approved the final version of the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical statement
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient’s parents and a legally authorized representative(s) for anonymized patient information and images to be published in this article.