
Abstract
A 15-year-old girl presented with new onset tonic-clonic seizures, encephalopathy, abdominal pain, and hypertension with a history of weight loss and emesis. Brain magnetic resonance imaging scans showed diffuse, bilateral cortical and subcortical gray and white matter signal abnormalities. Electroencephalography showed background slowing and disorganization. Extensive evaluation for infection, toxic metabolic, autoimmune disorders, and vasculitis were negative. She was noted to have dark red-colored urine with no red blood cells. Her urine porphobilinogen level was markedly elevated, consistent with the diagnosis of acute intermittent porphyria. She was treated with intravenous hemin with resolution of her neurologic and gastrointestinal symptoms. A hydroxymethylbilane synthase (porphobilinogen deaminase) pathogenic variant was found in porphyria gene panel confirming the diagnosis of acute intermittent porphyria. This case demonstrates a diagnostic challenge posited by a presentation of new onset seizure and encephalopathy due to acute intermittent porphyria, a rare and often overlooked condition in pediatrics.
Introduction
Acute intermittent porphyria (AIP) is an inherited disorder caused by a deficiency of hydroxymethylbilane synthase (HMBS, also known as porphobilinogen deaminase) in the heme biosynthesis pathway. 1 This deficiency leads to a buildup of heme precursors like 5-aminolaevulinic acid (ALA) and porphobilinogen. 2 AIP can present with a pattern of acute abdominal pain alongside autonomic and neuropsychiatric manifestations. 2 These can include a peripheral neuropathy with generalized paresthesia and muscle weakness or an encephalopathy with concomitant seizures, anxiety, agitation, and altered consciousness, with a manifestation consistent with posterior reversible encephalopathy syndrome (PRES).3,4 Cases involving neuropathy can also include autonomic dysfunction and acute polyneuropathy. Porphyria attacks have been documented usually around the age of puberty, with attacks noted to be extremely rare before the onset of puberty. 5 Accordingly, the incidence of neurological manifestations of AIP appears to be more pronounced in adults, 6 with limited literature available in children and adolescents. A long delay in diagnosis may prevent earlier identification around the onset of puberty. Hormonal changes during adolescence are one of the factors triggering attacks, as liver metabolism changes dramatically during adolescence and the menstrual cycle. Other triggers for acute attacks include infection, stress, fasting, smoking, or alcohol consumption. 2 Here, we report a rare case of a 15-year-old girl presenting with acute onset of seizures and mental status changes who was ultimately diagnosed with encephalopathy secondary to AIP.
Case presentation
A 15-year-old adolescent girl presented to our hospital with new-onset seizures. Parents reported difficulty waking her that morning, confusion and en route to the hospital, she had an episode where she was unresponsive with stiffening of her extremities. On the initial exam, the patient was hypertensive (147/92), emaciated, anxious, and confused with diffuse, sharp abdominal pain. The patient reported worsened abdominal pain in the last 7 days with intractable vomiting and minimal oral intake.
The patient was started on intravenous ondansetron and maintenance fluids. Complete blood cell count and complete metabolic panel labs were obtained early in the hospitalization and significant values are reported in Table 1. On admission, the patient reported she had just completed her menstrual cycle and endorsed smoking marijuana and vaping nicotine. Growth chart tracking revealed that she had lost 62 pounds over the course of 2 years, with a current body mass index of 17.14 kg/m2. Patient denied any prior history of seizures. Six hours into the admission, our patient experienced a focal onset bilateral tonic-clonic seizure. This episode started with stiffening in the right lower extremity that transitioned to whole body shaking for 3 min. Levetiracetam was started. On video electroencephalography, no seizures or epileptiform discharges, but background slowing, and disorganization were noted, suggestive of mild diffuse cerebral dysfunction and possible posterior focal dysfunction.
Significant laboratory values.

AST: aspartate aminotransferase; ALT: alanine transaminase.
Magnetic resonance imaging (MRI) of the brain was obtained, showing bilateral relatively symmetric areas of cortical signal within the frontal, parietal, and occipital lobes that was concerning for possible vasculitis, encephalitis, or hypertensive encephalopathy (see Figure 1).
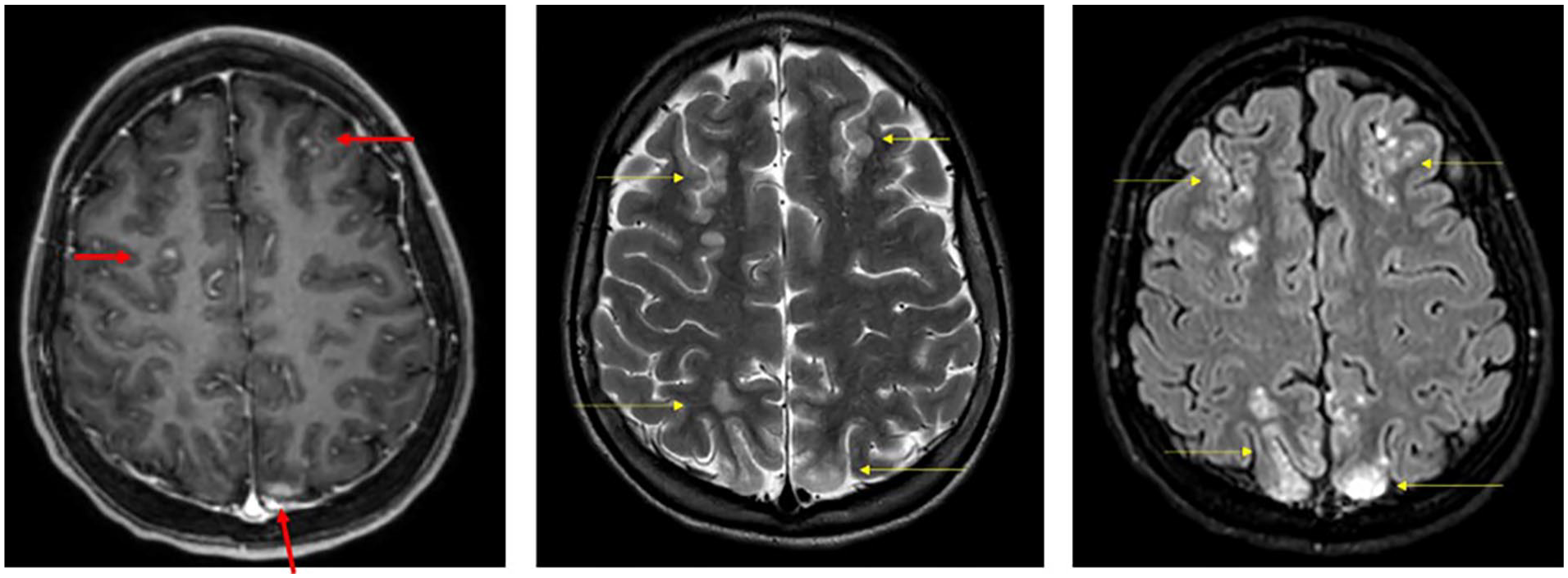
Magnetic resonance images of brain from initial presentation to the hospital. (left) Axial T1 post-contrast, (center) axial T2 weighted, and (right) axial Flair post-contrast. Images demonstrate Flair/T2 signal hyperintensity (yellow arrows) involving the cortex and subcortical white matter with patchy post-contrast enhancement (red arrows) involving the frontal, parietal, and occipital lobes in a watershed pattern (occipital lobe involvement not pictured here).
Due to concern for autoimmune encephalitis, our patient was started empirically on a 5-day course of methylprednisolone 20 mg/kg followed by two doses of intravenous immunoglobulin (IVIG) (1 g/kg). Cerebrospinal fluid (CSF) examination was remarkable only for the presence of two oligoclonal bands. Additional negative work up included tests for viral infections, tuberculosis, autoimmune encephalitis, hepatitis, vasculitis, and connective tissue disorders.
Due to persistent and severe abdominal pain and concern for constipation, the patient was placed on a bowel regimen. Given her recent history of marijuana use, she was treated for possible cannabis hyperemesis syndrome with capsaicin cream, acetaminophen, and ondansetron. Because she was unable to tolerate feeds, she was started on total parenteral nutrition on day 9 of admission, and this was continued until her abdominal pain and emesis improved, and she was taking enough calories orally to gain weight. Nasogastric feeds were attempted but not tolerated due to the discomfort and anxiety it brought. Her hypertension persisted, with a maximum blood pressure of 160/94. Given her hypertension and concern for possible hypertensive encephalopathy, she was started on anti-hypertensive therapy. She was placed on a fluid restriction due to her persistent hyponatremia (serum sodium ranging from 130 to 135 mmol/L) and possible syndrome of inappropriate antidiuretic hormone secretion (SIADH) given the presence of a high urine sodium (158 mmol/L) and a urine osmolality above 100 mOsm/L at 417 mOsm/L.
On day 5 of her hospitalization during her evaluation for persistent hypertension, she was noted to have a dark red-colored urine (Figure 2). Microscopic urinalysis revealed the absence of red blood cells. This unusual finding of dark red-colored urine without hematuria in addition to her neuropsychiatric symptoms and abdominal pain, led to the consideration of an acute porphyria attack as a possible overarching diagnosis. The patient’s random urine porphobilinogen level was markedly elevated at 314.5 µmol/L (normal <9 µmol/L). Other confirmatory testing included elevated total plasma/serum porphyrins (36 nmol/molCRT, normal = 0–15 nmol/L), urine uroporphyrin (87 µmol/molCRT, normal = 0–4 µmol/molCRT), urine heptacarboxylate (6 µmol/molCRT, normal = 0–2 µmol/molCRT), coproporphyrin I (10 µmol/molCRT, normal <30 nmol/molCRT), coproporphyrin II (83 µmol/molCRT, normal <30 nmol/molCRT), and urine ALA (158 µmol/L, normal = 0–35 µmol/L). Genetic testing subsequently revealed HMBS (C.510_511del, p.Asn173Hisfs*38) pathogenic variant confirming the diagnosis of AIP.

Image depicting the dark red-colored urine sample collected from patient. Urine tested positive for porphyrins.
Suspecting AIP, intravenous fluids containing dextrose, used as a treatment for AIP, 2 were reinitiated to diminish excess excretion of heme precursors. The patient continued to have hypertension, hyponatremia, and abdominal discomfort despite intravenous fluids, so on the 10th day of admission therapy was initiated with intravenous hemin at 4 mg/kg daily via central line access. Within 2 days of starting hemin, the patient began reporting improvement in her symptoms. After completion of 4 days of hemin therapy, the patient no longer complained of abdominal discomfort, became more alert and started “acting like herself.” Repeat brain MRI was obtained after hemin treatment to assess the reversibility of encephalopathic changes. Posthemin imaging showed evolving brain parenchymal signal abnormality with more confluent predominantly subcortical white matter involvement, microhemorrhage, and few small areas of diffusion signal abnormality (Supplemental Figure S1). There was also a notable interval development of thin supratentorial and infratentorial subdural hematohygromas ranging from 2.4 mm to 3.7 mm in thickness. In addition to resolution of neuropsychiatric symptoms, our patient’s symptoms of abdominal pain, constipation, dark urine, and hyponatremia all resolved, and her appetite returned. Given the patient’s stable clinical status and overall symptom improvement along with repeat MRI showing stable findings, she was discharged home on amlodipine and levetiracetam. After discharge, the patient returned to clinic for follow-up including multidisciplinary management of her AIP.
Discussion
This report serves to identify AIP as a rare and often misdiagnosed cause of encephalopathy and neuropsychiatric symptoms in pediatric patients. There have been only 15 reported pediatric AIP cases over the past 30 years, and a vast majority of these cases involved male patients. 7 Our patient’s initial presentation was confusing and complex with seemingly unrelated symptoms, eventually coming together to form a unifying diagnosis.
Among our initial differential for this girl with new-onset tonic-clonic seizures, we had entertained the possibility that this might be a case of anti-N-methyl-
However, our patient did meet the classic diagnostic triad of AIP: abdominal pain, neuropsychiatric symptoms, and reddish urine. 2 She also had many of the factors known to exacerbate AIP and place patients at risk for acute relapses. We suspect that our patient’s acute porphyria exacerbation was triggered by her vaping nicotine and concomitant tetrahydrocannabinol (THC) use, stress, weight loss, and menses. It is unclear exactly how nicotine and THC use affect heme biosynthesis. THC use may be involved in the induction of cannabis hyperemesis syndrome, which is characterized by bouts of episodic nausea and vomiting that can be worsened by cannabis intake. 12 However, THC use has been suggested as an appetite stimulant, 13 which may have a protective effect against AIP. Attacks of AIP appear more common after the onset of puberty in women, 5 possibly due to an effect from female sex hormones like estrogen and progesterone affecting liver metabolism. Similarly, weight loss and nicotine may contribute to changes in liver metabolism and stimulate heme biosynthesis.
The clinical presentation of our patient was similar to those reported in other cases of porphyric encephalopathy in the literature. 14 The most common presenting symptoms include seizures, confusion, lethargy, neuropathy, and visual dysfunction. Our patient had no acute signs of porphyria-related neuropathy; thus, we did not complete nerve conduction or electromyography studies. Additionally, she did not have any observable visual dysfunction, which are common in hypertensive crises and linked to edema in the posterior lobes. Prior to this report, only two pediatric patients, both pre-pubertal, were reported to have porphyric encephalopathy with MRI findings.14,15 Imaging findings in our patient were compatible with a PRES-like hypertensive encephalopathy reported in adults with porphyric encephalopathy. Intriguingly, after hemin therapy there was an interval development of thin subdural hemorrhages, an unexpected and unusual finding.
Patients with porphyric encephalopathy often show some improvement with adequate nutrition and fluids. The use of fluids to treat SIADH and for renal protection along with glucose for a patient in a fasted, dehydrated state with limited oral intake were helpful in this case, but not adequate for total symptom resolution. IV hemin is thought to show complete and faster resolution of symptoms.6,16 Consistent with the proven efficacy of IV hemin therapy, our patient improved significantly following hemin administration. 2 We did not observe resolution of lesions on MRI despite symptom resolution with treatment as we may have imaged too soon to see the full recovery on brain imaging. 17 Hemin therapy has been utilized prophylactically to prevent further exacerbations of AIP which may be a future consideration for our patient.18,19 Importantly, identification of precipitating factors and optimizing nutrition are important for preventing further attacks, including avoidance of fasted states and use of possible drug triggers like nicotine in our patient.2,3
Seizure prevention in AIP can be achieved with several anti-seizure medications (ASM) known not to exacerbate AIP including lamotrigine, levetiracetam, and lorazepam. 2 Levetiracetam is the drug of choice for seizures associated with porphyric encephalopathy while barbiturates and carbamazepine should be avoided.2,20 In general, the list of medications to avoid in patients with AIP can be obtained through the drug database (https://www.drugs-porphyria.org/). The length of long-term ASM therapy will depend on the risk of AIP recurrence. However, seizures generally do not present during the postattack period and may not necessitate the long-term use of an ASM.
Official porphyria guidelines from the Porphyrias Consortium of the National Institutes of Health’s Rare Diseases Clinical Research Network include (1) obtaining initial assessments for diagnostic confirmation by biochemical testing along with subsequent genetic testing to determine the specific acute hepatic porphyria, (2) counseling patients with a new diagnosis about avoiding known precipitating factors, (3) follow-up contingent on frequency of recurrent attacks. 21 Given the nature of the complexity of the disorder, AIP should be treated with a multidisciplinary approach to reduce morbidity and improve quality of life.
Conclusions
We hope to raise awareness of AIP in the pediatric population so that clinicians consider this diagnosis especially in cases of encephalopathy of an unclear etiology. It is critical for pediatric providers to correctly and quickly recognize, diagnose, and treat porphyric encephalopathy in children, particularly adolescent female patients presenting with abdominal pain and neuropsychiatric symptoms. Importantly, porphyria-directed treatment should not be started without the appropriate biochemical testing needed to diagnose the condition. In addition, it is very important to obtain genetic screening of the families (performed during follow-up in our case) in order to find patients at risk. This screening in families of patients with acute porphyria should be done before children reach puberty to help diagnose symptoms and signs of acute porphyria quickly and correctly.
AIP can have long-term health consequences if left undiagnosed and has increased mortality especially in patients with severe clinical manifestations requiring hospital admission. However, if patients are diagnosed in a timely fashion, outcomes are improved with appropriate management of acute symptoms and prevention of additional exacerbations. Importantly, this diagnostic process must include the recognition of acute porphyria on the differential when presented with the appropriate constellation of symptoms. In addition, appropriate follow-up is needed to ensure optimal quality of life for patients diagnosed with the condition.
Supplemental Material
sj-jpg-1-sco-10.1177_2050313X241298532 – Supplemental material for Porphyric encephalopathy in a 15-year-old girl: A case report
Supplemental material, sj-jpg-1-sco-10.1177_2050313X241298532 for Porphyric encephalopathy in a 15-year-old girl: A case report by Saihari S Dukkipati, Jordan Terschluse, Drew Thodeson, Angela Beavers, Melissa Muff-Luett, Claire Ives and Sookyong Koh in SAGE Open Medical Case Reports
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
The authors have obtained written informed consent from the patient’s mother. The signed consent form is available upon request. We confirm that Ethical Committee approval was not necessary. We confirm that guidelines on patient consent have been met and any details of informed consent obtained are indicated within the text of the submitted manuscript. Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.