
Abstract
We report the case of a patient who exhibits a concurrent diagnosis of type 1 diabetes mellitus, Gitelman syndrome and Cacci-Ricci disease. A 27-year-old male patient was diagnosed with Gitelman syndrome at the age of 3 years. Fourteen years later, he developed an autoantibody-negative type 1 diabetes mellitus. Cacci-Ricci’s disease was revealed by terminal hematuria and considered in view of the appearance found on the computed tomography (CT) scan. The finger-prick blood glucose level was 6 g/dl with no acetonuria. Creatinine clearance was 60 ml/min. Thyroid function tests were normal. Calcium, phosphorus and parathormone (PTH) levels were normal. Discussion: Gitelman syndrome is a rare disorder. The association between Gitelman syndrome and type 1 diabetes mellitus has been reported in the literature in two patients. Authors have investigated the association between Gitelman syndrome and type 2 diabetes mellitus. Several pathophysiological explanations have been put forward. Cacci-ricci disease is a rare, benign congenital anomaly. No association between type 1 diabetes mellitus, Gitelman syndrome and Cacci-Ricci disease has been reported in the literature. To our knowledge, this is the first case described in the literature.
Introduction
The human body is a complex network of interconnected systems where, sometimes, medical conditions can overlap or coexist in unexpected ways. Such is the case of a rare occurrence, where a patient exhibits a concurrent diagnosis of type 1 diabetes mellitus (T1DM), which is known to be associated with other autoimmune diseases, Gitelman syndrome, the most common hereditary tubulopathy 1 and Cacci-Ricci disease. This article explores the intriguing association between these three conditions and sheds light on their underlying mechanism.
Case presentation
A 27-year-old male patient, with no known family history, in particular no parental consanguinity or autoimmune diseases, was suspected to have Gitelman syndrome at the age of 3 years in the presence of tetany crisis, abdominal pain, and severe hypokalemia at 1.4 mmol/L. The diagnosis was confirmed by genetic testing highlighting a mutation in the SLC12A3 gene that codes for the sodium-chloride co-transporter. Abdominal pain disappeared after potassium and magnesium supplementation and the repercussion of Gitelman’s syndrome was growth retardation. Fourteen years later, the patient developed spontaneous ketoacidosis revealing an autoantibody-negative T1DM. He had poor glycemic control although intensified insulin protocol. Cacci-Ricci’s disease was revealed by terminal hematuria. CT scan objectified precalicial canalicular ectasia with bilateral nephrocalcinosis and about 20 non-obstructing bilateral micro-nephrolithiasis (Figure 1).
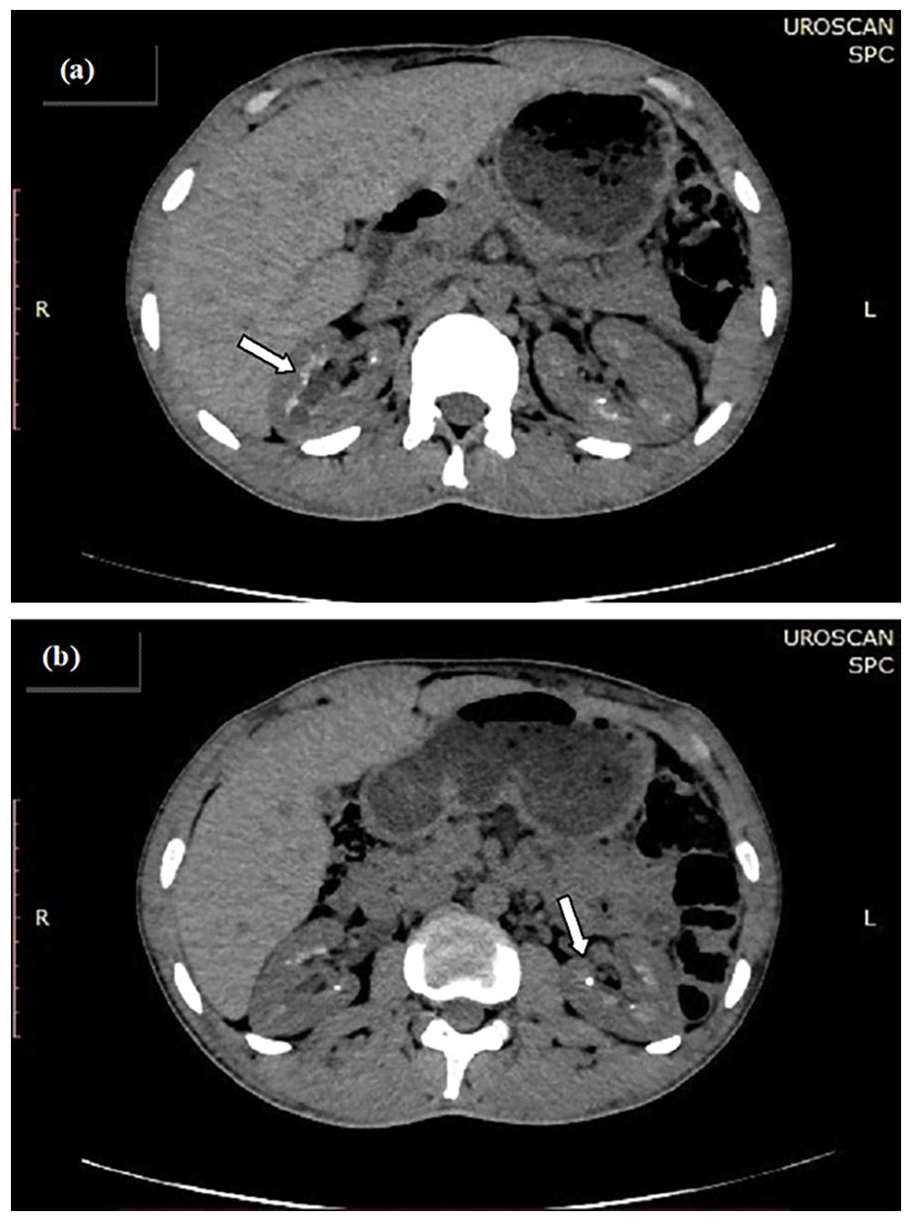
CT scan in a patient with type 1 diabetes mellitus, Gitelman syndrome and Cacci-Ricci disease showing the aspect of sponge kidneys (arrows), nephrolithiasis and nephrocalcinosis in the right (a) and left (b) kidney.
On presentation, the patient described asthenia and polyuro-polydipisic syndrome. He had no paresthesias or nephritic colic. Clinical examination revealed a body mass index of 21.4 kg/m2, a finger-prick blood glucose level of 6 g/dl with no acetonuria on urine dipstick, and correct blood pressure. The electrocardiogram was normal. The level of glycated hemoglobin was high, at 13%, testifying to chronic poor glycemic control. Creatinine clearance was 60 ml/min and microalbuminuria was negative. Thyroid function tests were normal. Serum calcium, phosphorus and PTH levels were in the normal range (Table 1).
Blood and urine biological parameters.

The patient is currently on potassium and magnesium supplements with clinical improvement. He is on intensified insulin therapy but still has uncontrolled diabetes with alternating hypoglycemia and hyperglycemia.
Discussion
The coexistence of T1DM, Gitelman syndrome and Cacci-Ricci disease in the same patient remains a fascinating medical mystery. Gitelman syndrome is a rare disorder. It is the most common hereditary tubulopathy. It generally presents in early childhood but can appear at a later age. 1 Gitelman syndrome is secondary to an inactivating mutation of the SLC12A3 gene encoding the sodium-chloride co-transporter of the distal convoluted tubule and is inherited in an autosomal recessive mode. 2 The pathophysiology of Gitelman syndrome has been extensively studied. The inactivating mutation of the SLC12A3 gene leads to dysfunction of the sodium-chloride co-transporter, causing sodium leakage and mimicking the use of thiazide diuretics. Hyperaldosteronism secondary to sodium depletion and hypovolemia causes hypokalemia and metabolic alkalosis. This phenomenon is associated with inhibition of the TRPM6 channel, leading to urinary magnesium leakage and hypomagnesemia, which in turn inhibits PTH secretion, resulting in hypocalcemia and hypocalciuria. 3 Gitelman syndrome can be the cause of growth retardation if the disease is revealed in childhood,1,4 as was the case with our patient. It may be associated with pubertal delay and cardiac disorders due to hypokalemia and hypomagnesemia. Chondrocalcinosis may be seen as a consequence of hypomagnesemia, and renal function may rarely be impaired due to secondary hyperaldosteronism. 4
The association between Gitelman syndrome and T1DM has been reported in the literature in two patients. The first patient was a 14-year-old boy with T1DM since the age of 9 and Gitelman syndrome since the age of 5. He was hospitalized for the management of diabetic ketoacidosis precipitated by insulin discontinuation, in which hypokalemia at 2.2 mmol/L was objectified. 5 The second patient was a 10-year-old Tunisian boy who was hospitalized for vomiting, weight loss and asthenia. He was diagnosed with ketoacidosis revealing diabetes mellitus where severe hypokalemia at 1.1 mmol/L was observed. Genetic studies confirmed a homozygous mutation of the SLC12A3 gene in the boy and a heterozygous mutation in the parents. 6
Other authors have investigated the association between Gitelman syndrome and type 2 diabetes mellitus and many cases have been reported.7–9 Several pathophysiological explanations have been put forward, involving hypokalemia, hypomagenemia and secondary hyperaldosteronism. 8 Indeed, hypokalemia prevents the closure of adenosine triphosphate (ATP)-sensitive potassium channels on the surface of pancreatic β-cells, thereby inhibiting insulin secretion, and can lead to β-cell dysfunction and even apoptosis. Hypomagnesemia, on the other hand, reduces tyrosine kinase activity at insulin receptors and disrupts ATP-dependent potassium channels and L-type calcium channels in β-cells, thereby decreasing insulin sensitivity and reducing insulin secretion. Secondary hyperaldosteronism stimulates endothelial remodeling and decreases insulin supply to target cells, as well as insulin resistance in various tissues and organs, including adipose tissue and the liver.
Cacci-Ricci or sponge kidney disease is a rare, benign congenital anomaly with an estimated prevalence of 1/5000. 10 The age of onset is generally between the second and third decade of life, although cases have been reported in newborns. 11 The main anomaly in Cacci-Ricci disease is dilatation of the medullary and papillary portions of the collecting ducts. Generally asymptomatic, it may present as hematuria, recurrent urinary tract infections or kidney stone formation. Renal involvement is bilateral in 70% of cases. 10 Although it is sporadic most of the time, 5% of cases are hereditary, with autosomal dominant inheritance. The causative mutation concerns the receptor tyrosine kinase (RET) gene and, more rarely, the glial cell line-derived neurotrophic factor gene. 12 Cacci-Ricci disease can result in nephrolithiasis and nephrocalcinosis, 10 both of which were found in our patient. Urinary tract infections are also a common complication of this disease. 10 The osteopenia or osteoporosis seen in Cacci-Ricci disease can be explained by increased calcium excretion, altered urinary acidification and hyperparathyroidism, which may be associated with the disease in some patients. 13
An association between Cacci-Ricci disease and bilateral pheochromocytoma has been described in the literature in a 46-year-old man. 14 The thoraco-abdomino-pelvic CT scan revealed a 7 × 7.5 cm left adrenal mass and a 9 × 8 cm right adrenal mass with sponge kidneys and a pituitary adenoma. The thyroid and parathyroid glands were without abnormalities. Pathological examination revealed bilateral pheochromocytoma. Genetic analysis identified the presence of a RET proto-oncogene mutation. In fact, the RET gene is involved in kidney formation during embryogenesis. Therefore, mutations in the RET gene can lead to renal dysplasia. 12
No association between T1DM, Gitelman syndrome and Cacci-Ricci disease has been reported in the literature.
Conclusion
This case illustrates a fascinating association between three different and independent pathologies. To our knowledge, it is the first case described in the literature. To date, no pathophysiological explanation has been put forward to explain this association. As research continues, we hope to better understand the underlying mechanisms and develop more effective treatment strategies for these rare and complex cases.
Footnotes
Acknowledgements
The abstract available on JCEM Case Report.
Authorship contributions
The authors gratefully acknowledge the patients who participated in this study and thank M.B.B. for the conception of the manuscript, F.B.A. and A.G. for help with referencing, H.E. for the supervision and Y.H. and M.C. for their guidance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.