
Abstract
We present the cases of two brothers with ichthyosis, born to consanguineous parents, with the eldest having extracutaneous manifestations in the form of microphthalmia and corneal opacities causing complete blindness. Initially, we were faced with the question of whether the phenotype in this family was due to the effects of a single pleiotropic, presumably autosomal recessive gene manifesting as a syndromic form of ichthyosis, or whether there were multiple causal genes, and the ichthyosis was non-syndromic. Ultimately, clinical follow-up of the family, combined with research-based exome sequencing established a diagnosis of NIPAL4 autosomal recessive congenital ichthyosis in both brothers, but the ocular abnormalities causing blindness in the older brother were due to coexisting autosomal recessively inherited loss of function mutations in peroxidasin, the latter finding also seen in a sister unaffected by ichthyosis.
Case reports
Patient 1
Patient 1 was seen by genetics at 7 days of age, as he presented with microphthalmia and dry skin. At age 4.5 months, during an admission to paediatric ICU for a viral illness, he demonstrated ichthyotic skin changes. At 9 years of age, he was referred to our dermatology clinic, and the clinical diagnosis of ichthyosis was confirmed with the presence of dark thickened scales over his neck, and extremities including the flexor surfaces of antecubital and popliteal fossae. Palms showed mild hyperlinearity but no keratoderma, and hair, teeth and nails were normal. At birth, he had normal appearing skin with no collodion membrane. In addition to ichthyosis, his congenital microphthalmia and subsequent corneal opacities had led to complete blindness. There was a history of cryptorchidism but no history of delayed labour at birth.
Patient 2
Patient 2, the younger brother of patient 1, was assessed in our dermatology clinic at 4 years of age with a history of dry skin starting at 8 months. He was diagnosed with ichthyosis, appearing as diffuse generalized dark thickened scales on his body but no keratoderma. Like his older brother, he had a history of normal skin at birth with no collodion membrane and no history of delayed labour at birth. However, he differed from his brother, as he did not present with any ocular abnormalities or a history of cryptorchidism. In addition, he was diagnosed with a carnitine uptake deficiency diagnosed on neonatal metabolic screening necessitating ongoing treatment with oral L-carnitine.
The brothers are from a Kurdish family with five children and consanguineous parents. None of the other three children have ichthyosis. However, one of the female siblings has microphthalmia leading to corneal opacities and blindness similar to her older brother. The other two female siblings are healthy (Figure 1).

Pedigree of the family of patients 1 and 2 demonstrating consanguinity, and clinical presentations of the five siblings. Numeric labels are in order of birth. Case report: patient 1 is labelled (1) and patient 2 is labelled (4) within the pedigree.
This family was previously briefly reported as Family #6 in a genetics journal, 1 the purpose of which was to emphasize the use of Whole Exome Sequencing (WES) to diagnose multiple genetic diagnoses in probands, and their families referred for analysis in two national research programmes in Canada (FORGE and Care4Rare Canada). This publication did not discuss the dermatologic features or the diagnostic dilemma when faced with patients with ichthyosis and ocular abnormalities in detail, which is the major reason for this publication.
Genetic testing and investigations
Early genetic testing on patient 1 consisted of a normal karyotype and a normal fluorescence in situ hybridization using a steroid sulfatase probe. A skin biopsy was obtained for cultured fibroblasts and showed normal enzyme activity for fatty aldehyde dehydrogenase and arylsulfatase.
The family was eventually enrolled in a research-based exome sequencing project (FORGE Canada), when patient 1 was age 14 and patient 2 was age 7, showing in both patients, a novel homozygous likely pathogenic frameshift variant in the NIPAL4 (ichthyin) gene, specifically c.1310_1317del [p.Leu437Glnfs*35], leading to the diagnosis of non-syndromic NIPAL4 ARCI (OMIM 612281) and clinically they both had lamellar ichthyosis-like features. In addition to this mutation, patient 1 was also found to have a rare (present twice in the heterozygous state in gnomAD) homozygous likely pathogenic missense variant in the PXDN gene (c.3211A>G [p.Arg1071Gly]), responsible for his symptoms of microphthalmia and corneal opacities (OMIM 269400), whereas patient 2 had a co-existing homozygous variant in the SLC22A5/OCTN2 gene (c.95A>G [p.Asn32Ser]) responsible for a carnitine uptake defect (OMIM 212140).
Discussion
The ichthyoses are a group of inherited Mendelian disorders of cornification affecting all or most of the integument. Patients with inherited ichthyoses present with hyperkeratosis and scaling and varying degrees of erythema. 1 Inherited ichthyoses can be syndromic, with extracutaneous manifestations as well as non-syndromic 2 that will present primarily with cutaneous manifestations. ARCI, a non-syndromic form of ichthyosis, has been reported to be caused by 14 different genes 3 with the most common being TGM1, NIPAL4 4 and ABCA12. Three large patient series have confirmed NIPAL4 as the second commonest cause of ARCI.3,5,6 The majority of NIPAL4-related ichthyosis will present as congenital ichthyosiform erythroderma-like, 7 however, a lamellar ichthyosis-like phenotype can also occur. 7
Patients can be born with a collodion membrane occurring in 28% in an English cohort. 3 A yellowish diffuse palmoplantar keratoderma was suggested as a phenotypic clue to NIPAL4 ichthyosis. 8 This was not supported in other case series. 3
When faced with only males in a family with ichthyosis, X-linked ichthyosis (XLI) is often the first consideration as it almost exclusively occurs in males, and most often appears as brown scales on the neck, trunk, and lower extremities with flexural involvement. 9 In our two brothers, it was originally difficult to determine whether ichthyosis was an isolated issue, or part of a syndrome. Patient 1’s clinical presentations were suggestive of XLI, as he presented with corneal opacities and cryptorchidism, whereas his younger brother presented solely with ichthyosis and no ocular abnormalities. However, XLI does not cause blindness, and the presence of similar microphthalmia and corneal opacities in the second oldest sister suggested that the blindness was likely not associated with the inherited ichthyosis that occurred in the two brothers.
WES testing finally confirmed our suspicion that the presentation of ichthyosis, microphthalmia and corneal opacities were due to two separate autosomal recessive gene mutations, and not part of a syndromic ichthyosis with ocular involvement, such as XLI, 9 ichthyosis–follicularis–atrichia–photophobia (IFAP), 10 Sjogren–Larsson syndrome, 11 Senter–KID (keratitis, ichthyosis and deafness) syndrome 12 or a single case report of a family with microphthalmia and ichthyosis reported by Loffredo et al. 13
WES identified that the ichthyosis in both patients was due to a homozygous NIPAL4 variant. The ocular symptoms in patient 1 and his sister were due to a separate variant in the PXDN gene 14 demonstrating the complexity of clinically diagnosing inherited genetic conditions such as ichthyosis with potential syndromic features in consanguineous families, in whom multiple coexisting autosomal recessive conditions can occur in the same patient at a higher frequency. 1
The first-line treatment for ichthyosis is using topical emollients and keratolytics such as urea, alpha-hydroxy acids, propylene glycol or salicylic acid. 15 Further management can include systemic retinoids such as acitretin at doses of 0.2–0.5 mg/kg/day. 16 Both our patients have used multiple moisturizers as well as 20% urea cream without much improvement. Finally, oral acitretin 10 mg daily was prescribed for each of the two brothers, resulting in significant improvement of their ichthyosis (Figures 2 and 3).

(a) Patient 1 presenting with ichthyosis demonstrating dark thickened scales on his chest. (b) Patient 1 demonstrating clearance of ichthyosis at 2 months following treatment with oral acitretin 10 mg daily.
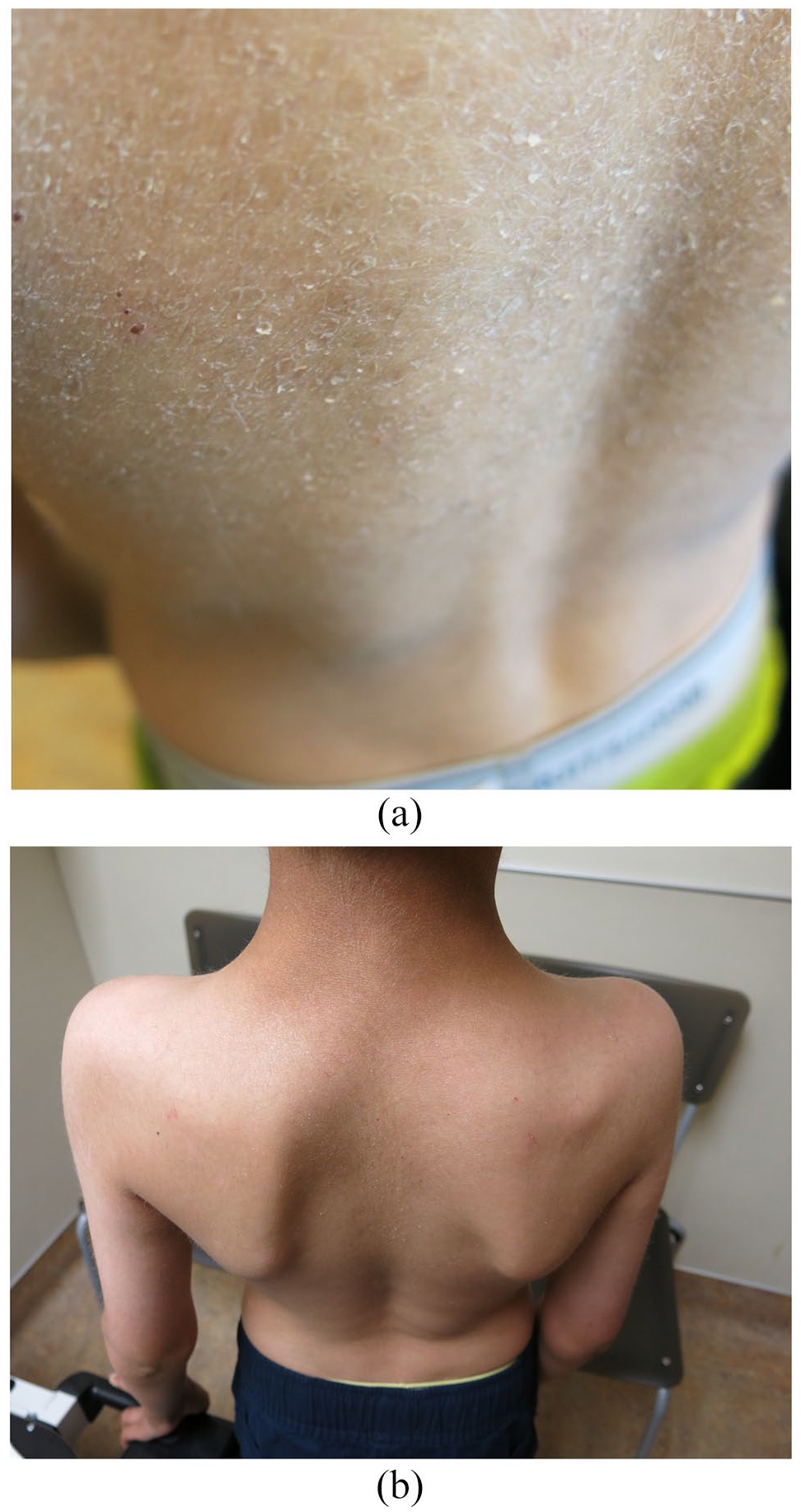
(a) Patient 2 presenting with ichthyosis demonstrating dark thickened scales on his back. (b) Patient 2 demonstrating clearance of ichthyosis at 2 months following treatment with oral acitretin 10 mg daily.
Footnotes
Consent
We obtained consent from the patients referenced in this publication including permission to publish their relevant medical information and photographs.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.