
Abstract
Sirenomelia is a rare lethal multi-systemic birth malformation in which the two lower limbs are replaced with a rotated single midline tail-like limb. Several hypotheses try to explain this syndrome, with the most prominent theories being the “vascular steal hypothesis” and the “defective blastogenesis hypothesis.” We report a case of a baby with sirenomelia who had a single femur and a single tibia, which classify the case as type VI on Stocker and Heifetz classification. The only risk factor in our case is young maternal age. The baby had a single umbilical artery, a prominent feature of the vascular steal hypothesis. Nonetheless, it also had upper limb deformity, which can be better explained by the defective blastogenesis hypothesis. Our case supports the defective blastogenesis theory of sirenomelia more than the vascular steal hypothesis as it has both a single umbilical artery and upper limb deformity. Also, our case serves as a teaching lesson that indicates the importance of an obstetric ultrasound before a cesarean section has to be done to avoid unnecessary surgery for life incompatible congenital anomaly.
Keywords
Introduction
Depicted as half woman, half fish, Atargatis, the Syrian goddess will always be a reminder of a forgotten time when birth defects catalyzed human imagination. 1 Sirenomelia is a rare lethal multi-systemic birth malformation in which the two lower limbs are replaced with a rotated single midline tail-like limb.2,3 Other features of the syndrome explain its high mortality rate, the most important of which is renal agenesis and lung hypoplasia. 4 Older studies found the prevalence of sirenomelia to be anywhere between 1.03:100,000 and 1.19:100,000, 2 but newer studies have lower estimations at 0.98:100,000. 3 Multiple risk factors have been linked to the syndrome, including young maternal age,2,3 maternal diabetes, and twinning.2,4 Multiple scales estimate the severity of the malformation, most of which correlate the severity of the syndrome with the degree of absent foot structures. 4 The classification created by Stocker and Heifetz remains widely used to describe the anomaly. 5 It classifies the syndrome into separate groups based on the skeletal structure of the single limb. 5 Diagnosing this syndrome poses a challenge as renal agenesis leads to severe oligohydramnios but it can be diagnosed between 8 and 16 weeks of gestation since the production of the amniotic fluid is mainly maternal at this point in pregnancy. 3 Several hypotheses try to explain this syndrome but none have accumulated enough scientific evidence, with the most prominent theories being the “vascular steal hypothesis” and the “defective blastogenesis hypothesis.” 4 Recent hypotheses suggest a connection between caudal dysgenesis and sirenomelia, classifying them as part of one spectrum. 6 Here, we present a type VI case on Stocker and Heifetz classification, with a non-diabetic mother aged 18, that supports the defective blastogenesis hypothesis. The syndrome was not diagnosed until birth.
Case presentation
An 18-year-old Syrian primigravida at 30 weeks of gestation presented in the birthing unit, complaining of regular uterine contractions. She mentioned that she had noticed a sudden gush of copious vaginal discharge 2 days prior, which was suspected to be a rupture of membranes. There was no history of smoking or medications during pregnancy, nor was there any history of intrauterine infections. Consanguineous marriage was denied, and there was no history of genetic disorders or familial malformations. The patient is from Rastan in Homs which was severely affected by war. The mother’s standard blood tests were all within the normal range. Severe oligohydramnios was recorded, and preterm labor was diagnosed based on cervical changes and active labor. Rupture of membranes and non-reassuring fetal status were considered contraindications to tocolysis. A decision to terminate the pregnancy by labor induction was made. Thus, a single course of betamethasone was administered to stimulate fetal type II pneumocyte surfactant production, and intravenous magnesium sulfate was given to protect the fetus from cerebral palsy. Then, labor induction was obtained with oxytocin. Continuous cardiotocography showed irregular fetal heart rate which did not respond to resuscitation procedures so an urgent lower segment cesarean section was performed, resulting in a premature baby with sirenomelia. Sirenomelia was not detected until birth due to a lack of regular check-ups during pregnancy and severe oligohydramnios at the time of presence. The baby was intubated and put on a breathing circuit as spontaneous breathing failed. The baby had ambiguous genitalia, a single umbilical artery, Potter syndrome facial characteristics (low-set ears, flat nose, and chin), skin tags parallel to their sacral vertebrae (see Figure 1), left radial absence (see Figure 2), and most importantly, a single lower limb (see Figure 1). A computed tomography (CT) scan showed a single femur and a single tibia, without other skeletal components, which classified the case as a type VI on Stocker and Heifetz classification (CT scan video is available in the Supplemental Material). The right lung collapsed, and both lungs were hypoplastic, as evidenced by the bell-shaped thorax on the X-ray image (see Figure 2). Lack of amniotic fluid can cause both pneumothorax and pulmonary hypoplasia. The transverse colon was enlarged, corresponding with an imperforate anus (see Figures 1 and 2). The CT was done without contrast, so the status of the kidneys could not be confirmed, but they did not appear normal, and the severe oligohydramnios confirmed renal dysfunctionality. The baby passed away a few hours after birth. An autopsy of the fetus was not performed due to limited availability, high costs, and the parents’ refusal.

(a) Potter syndrome facial characteristics with a flat nose and chin. (b) Skin tags parallel to sacral vertebrae and an imperforate anus. (c) Sirenomelia phenotype type VI case on Stocker and Heifetz classification with Potter syndrome facial characteristics, including low-set ears.
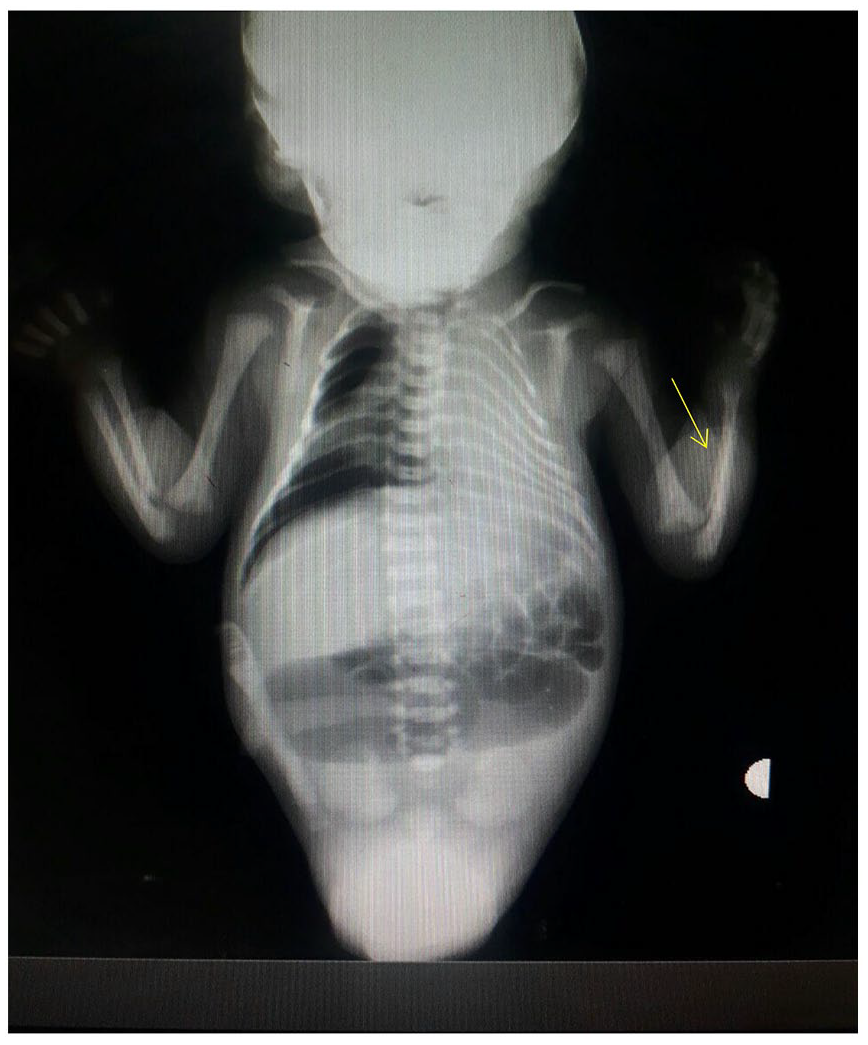
An anteroposterior X-ray image showing left radial absence (the yellow arrow), collapsed right lung, significant right pneumothorax, and bilateral lung hypoplasia, indicated by the bell-shaped thorax. The enlarged transverse colon corresponds to an imperforate anus.
Later, after a second pregnancy, the patient had a normal infant.
Discussion
Studies have reported sirenomelia diagnoses as early as the eighth week of gestation on prenatal ultrasound. 3 As pregnancy progresses, lower amniotic fluid levels decrease the accuracy of this method.2,3 In our case, the mother did not get regular ultrasound check-ups, and the pregnancy continued. This emphasizes the importance of regular check-ups.
Many risk factors for sirenomelia exist. Our case, however, has only one risk factor, which is, young maternal age. She is not diabetic, and no evidence of twinning exists. In Syria, marriage at a young age, which studies associate with different types of non-chromosomal birth defects, 7 is common. 8 Henceforth, there is a need for studies in Syria to evaluate the problem and for educational initiatives about the risks of pregnancy in teenage mothers as well.
Aside from the previously mentioned risk factors, several chemicals could induce sirenomelia in experimental mammals, such as retinoic acid, cadmium, lead, and ochratoxin A. 3 Chemical pollutants such as arsenic, lead, copper, and other heavy metals can pass through the placenta and can cause consequences to the mother and the child so that reproductive toxicology has considered them as teratogens. 9 Though evidence to associate teratogens with sirenomelia in humans is lacking. 3 It remains possible that our case was exposed to teratogens, due to war residues and the lack of proper general health education in Syrian society, 10 which calls for educational initiatives in this area.
In studying the syndrome, the research did not prove chromosome abnormality.2,4 Orioli et al. 3 reported no familial recurrence of sirenomelia. However, several cases show the familial occurrence of sirenomelia: a family had four cases of renal dysplasia, three siblings, and an aunt, while a fourth sibling suffered from sirenomelia. 4 In another family, a child had several pelvic malformations, whereas the second child had sirenomelia. 4 Familial recurrence of renal malformation and sirenomelia might provide evidence for sirenomelia as part of the caudal regression spectrum. Familial recurrence, if true, can be explained by exposure to certain teratogens because of the shared environment. 4 Pregnancies in developing countries tend to be exposed to potential teratogens to a higher degree than in industrialized nations. 10 The second explanation is a common genetic connection, which has not been proved yet. 4 Unfortunately, genetic mapping is not available for our case, and the next pregnancy resulted in a normal infant.
Two major pathogenic hypotheses try to explain the syndrome:
(1) A flaw in or damage to the caudal mesoderm is thought to develop at the tailbud stage, which corresponds to the third week of human pregnancy, according to the defective blastogenesis theory. 2 Cloacal and urogenital-derived malformations, as well as dysfunctional midline structures, are the outcomes. Thus, sirenomelia can be classified as a severe form of caudal regression spectrum.2,3,11,12 The underlying event’s intensity, onset, and duration all affect how variable the phenotype is Garrido-Allepuz et al. 12 In other words, during embryonal development, a single “hit” may result in abnormalities in many progenitor fields, leading to polytypic manifestations. The time and intensity of the hit determine the heterogeneity. 13
(2) The vascular steal phenomenon, where the distal aorta is atretic, and one intra-abdominal artery is present, leads to the redirection of blood from caudal structures to the placenta and results in anomalies of caudal structures.2,11 In 11 cases by Stevenson et al., a single large artery in the abdominal cavity was present, pushing the vascular steal theory forward.4,14 However, a single umbilical artery can also be found in some patients with caudal regression syndrome and normal umbilical arteries in some patients with sirenomelia.4,6 Our case is unique as it has a single umbilical artery, a prominent feature of the vascular steal hypothesis. Nonetheless, it has upper limb deformity, that is, left radial absence. The aforementioned means that our case is considered to support the defective blastogenesis hypothesis, as the vascular steel phenomenon cannot explain the cause of radial absence. It also suggests that deficient blastogenesis could concomitantly affect organ and vessel development as mentioned by Chikkannaiah et al. 13
Studies have reported occasional survivors. The phenotype varies in each malformation syndrome. Thus, few infants with sirenomelia, who have not had bilateral renal agenesis, have survived. 2 Because this sequence has a severe prognosis, the option to terminate the pregnancy should be offered to the parents before viability.15,16 But such an option is only possible in 39% of the world’s countries. 17
Finally, the importance of preventing congenital anomalies and genetic disorders is clear to everyone, especially in developing countries such as Syria. Here we include several recommendations by WHO: enhancement of epidemiological knowledge about genetic disorders and birth defects, improvement of pre-and perinatal services, including family planning and maternal nutrition, and education of health professionals and the public on genetics and birth defects. 10
Conclusion
Our case supports the defective blastogenesis theory of sirenomelia more than the vascular steal hypothesis as it has both a single umbilical artery and upper limb deformity. Also, our case serves as an educational example that highlights the significance of conducting an obstetric ultrasound prior to performing a cesarean section to prevent a medically futile surgery due to a non-viable fetal condition.
Supplemental Material
sj-docx-1-sco-10.1177_2050313X241229589 – Supplemental material for Defective blastogenesis of postnatally diagnosed type VI sirenomelia in a young primigravida: A case report
Supplemental material, sj-docx-1-sco-10.1177_2050313X241229589 for Defective blastogenesis of postnatally diagnosed type VI sirenomelia in a young primigravida: A case report by Mhd Mustafa Albitar, Mohamad Moamen Almouallem, Ahmad Mostafa Kanaan and Ieman Alawad in SAGE Open Medical Case Reports
Footnotes
Acknowledgements
We wish to express our appreciation to Stemosis for Scientific Research, a Syria-based scientific research youth association managed by Nafiza Martini, for the scientific environment they provided. We also thank Dr. Rafif Terkawi for her assistance with X-ray and CT image readings, Dr Eyad Abdullah for his assistance in collecting patient data, Dr Abdul Muttaleb Alsah for his scientific help, and Dr Fatima Alawad for her technical help.
Author contributions
M.M.Al. contributed to drafting, reviewing, and editing; M.M.Alm. contributed to drafting, reviewing, and editing; A.M.K. contributed to drafting, reviewing, and editing; I.A. contributed to data collecting, drafting, reviewing, editing, and supervising. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
As the study is based on both the mother and her infant, written informed consent was obtained from the mother for her information to be published in this article. In addition, written informed consent was obtained from both parents “the legally authorized representative of the baby” for their infant information to be published in this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.