
Abstract
Burnside–Butler syndrome is an inheritable genetic condition characterized by the partial deletion of specific genetic material located on chromosome 15q11. Individuals diagnosed with this particular medical condition display a variety of neuropsychiatric disorders, including psychosis, aggression, mood disorders, anxiety disorders, developmental disorders involving learning difficulties, language delays, autism spectrum disorders, and attention-deficit/hyperactivity disorder. The authors discuss the case of a 51-year-old Caucasian female diagnosed with Burnside–Butler syndrome at 8 years. The article highlights the importance of raising awareness regarding the complex nature and delayed onset of neuropsychiatric symptoms associated with this syndrome. It also emphasizes the need for comprehensive evaluation and multidisciplinary care for individuals affected by this uncommon condition.
Introduction
Burnside–Butler syndrome (BBS) is an infrequent genetic disorder characterized by developmental delays, congenital abnormalities, neurobehavioral issues (such as dyslexia, autism, and psychosis), and atypical brain imaging findings. It is caused by microdeletions on chromosome 15, in the proximal long arm segment 15q11-q13, which contains five breakpoint sites, notably BP1BP5.1,2 Owing to a cluster of low-copy DNA repeats in this region, homologous chromosome 15 pairs often misalign during meiosis, causing chromosomal anomalies.
There are two types of deletion that cause Prader–Willi syndrome (PWS) and Angelman syndrome (AS), which are genomic imprinting errors in humans. PWS involves deletions of the paternal chromosome 15q11-q13 region and is associated with more severe clinical impairments, whereas AS involves deletions of the maternal chromosome 15q11-q13 region.2,3 Individuals with PWS and larger type I deletions exhibit more severe clinical impairments than those with deletions specifically in the 15q11.2 BP1-BP2 region, as observed in individuals with BBS.
The 15q11.2 BP1-BP2 microdeletion is situated between BP1 and BP2 regions, including four genes: non-imprinted in Prader–Willi/Angelman syndrome region protein 1 (NIPA1), non-imprinted in Prader-–illi/Angelman syndrome region protein 2 (NIPA2), cytoplasmic FMR1-interacting protein 1 (CYFIP1), and tubulin gamma complex component 5 (TUBGCP5). Mutations in these genes lead to various clinical impairments, including hereditary spastic paraplegia, childhood absence epilepsy, attention-deficit hyperactivity disorder (ADHD), obsessive-compulsive disorder (OCD), and linkage to autism-related genes. BBS, being an autosomal dominant genetic disorder with incomplete penetrance, presents varied expression among people with this syndrome.1,3 Hence, not all individuals with microdeletions within the 15q11.2 band are clinically affected and may not exhibit the same symptoms and features. This case report highlights an unusual instance of late-onset psychosis in a patient with BBS and emphasizes the importance of comprehensive psychiatric evaluations for such individuals. The authors aim to contribute to increasing research into psychiatric manifestations of genetic syndromes.
Case report
A 51-year-old Caucasian female, accompanied by her caretaker, presented to the psychiatric hospital with complaints of active auditory hallucinations. There was a lack of information regarding the patient’s family history, and attempts to contact family members were unsuccessful. She was referred by her general practitioner due to a gradual decline in her mental health. This was despite her being reportedly compliant with her prescribed medications. Prior to her hospital presentation, the patient had already received a diagnosis of mild intellectual disability along with cerebral palsy, specifically exhibiting mild left-side hemiparesis. Genetic analysis with fluorescent in situ hybridization (FISH) 4 revealed a microdeletion at 15q11.2 and at BP1-BP2 at 18, which was noted in the history. However, the treating physician did not have access to her FISH reports from her previous hospital. The patient also experienced chronic dysthymia due to multiple psychosocial stressors in her early 20s. These stressors included losing custody of her child, dealing with her child with autism spectrum disorder (ASD), intimate partner violence, and sexual abuse.
The patient had been treated with 100 mg of desvenlafaxine for over 20 years to manage chronic dysthymia. Throughout this period, the patient’s condition remained relatively stable. However, over a period of 7–8 days, she began to exhibit signs of a thought disorder characterized by paranoia about her environment and altercations with family members. She complained of auditory hallucinations that commanded her to harm herself and others. Upon examination, her thought content was found to be delusional. She believed there was an entity inside her brain, capable of hearing all her thoughts. Her behavior declined, and she became primitive in nature. She also showed signs of disorganized thought processes, with loose associations and incoherence.
The patient and her caretaker provided negative reports regarding recent infections, fever, or any significant events. These reports could potentially have contributed to the sudden worsening of the patient's condition. The patient reportedly denied substance or alcohol abuse. A thorough medical and psychiatric assessment of the patient ruled out recent infection, trauma, substance abuse, history of trauma or fall, metabolic/electrolyte abnormalities, and adverse side effects of prescribed medicines. A CT scan was also performed to rule out potential neurological causes of late-onset psychosis. 5 As part of the treatment for her psychosis, the patient was prescribed oral olanzapine at a starting dose of 5 mg. This dose was gradually increased to 20 mg over 1 week of inpatient treatment. Within 2–3 days of initiating treatment, the patient exhibited significant improvement, eventually leading to her discharge after 1 week of treatment. The patient demonstrated insight into her condition and was oriented to time, place, person, and situation. During a subsequent follow-up held 2 weeks later, the patient’s condition was reported to be stable, and she demonstrated compliance with all prescribed medications. The patient continued to be followed up every 3 months for 2 years.
Discussion
In a comprehensive review conducted by Cox et al. 1 in 2015, the clinical features of 200 individuals with BBS were categorized, and their frequencies were reported. The categories included:
Developmental delays (73%) and speech delays (67%)
Dysmorphic ears (46%) and palatal abnormalities (46%)
Challenges in writing (60%), reading (57%), and memory (60%), as well as verbal IQ scores 6 below 75 (50%)
Unspecified behavioral issues (55%)
Abnormal brain imaging (42%)
Less frequent characteristics included epilepsy or seizures (26%), ASD (27%), ADHD (35%), and schizophrenia or paranoid psychosis (20%). 1
These effects, however, may not necessarily appear uniformly across all affected persons as they tend to show reduced penetrance along with variable phenotypic expressivity. 1 Incomplete penetrance (10.4%–11% for congenital anomalies, developmental delay, intellectual disability, and autism, and 2% for schizophrenia) may be related to subclinical neuropsychiatric manifestations or influenced by a lack of data on the parents of individuals with the microdeletion or control cohort participants. 1
The 15q11.2 BP1-BP2 microdeletion was reported to have a prevalence of 0.6%–1.3% among symptomatic individuals undergoing chromosomal microarray testing.1,2 According to Caffarkey et al., 2 51% of microdeletion carriers inherited the condition from apparently unaffected parents, while 35% did so from an affected parent. They also reported a de novo frequency ranging from 5% to 20%.
Davis et al. 3 identified variations in BBS clinical presentations depending on the deletion origin (maternal or paternal). Maternal origin deletions are associated with a higher risk of developmental, motor, and speech delays, intellectual and learning problems, autism, and behavioral/psychiatric diagnoses. Paternal origin deletions, on the other hand, are more strongly associated with poor coordination, ataxia, and congenital anomalies such as congenital heart disease. NIPA1, NIPA2, TUBGCP5, and CYFIP1 are four highly conserved and unimprinted protein-coding genes located in the 15q11.2 BP1-BP2 region.1,2,7–10 NIPA1 and NIPA2 encode magnesium transporters associated with spastic paraplegia and absence epilepsy, respectively.1,2,7–12 TUBGCP5 has been associated with ADHD and OCD. 9 Reduced expression of CYFIP1 has been linked to the disruption of gene pathways associated with schizophrenia.
Numerous studies have established significant associations between microdeletion and conditions such as schizophrenia and related psychoses.1,3,9,13–17 This is supported by the observation that people with learning disabilities and schizophrenia had a higher prevalence of the 15q11.2 deletion than the general population. 13 In schizophrenia patients, the prevalence of the 15q11.2 BP1-BP2 deletion ranges from 0.14% to 0.65%. 9 However, the presence of a particular copy number variant (CNV) alone is not sufficient evidence for it to be a causative factor 9 owing to its low penetrance (2% for schizophrenia), which supports the hypothesis that a combination of genetic and environmental factors are responsible for psychoses and schizophrenia.7,11,15 Furthermore, it has been reported that individuals diagnosed with schizophrenia and pathogenic CNVs have an increased likelihood of resistance to treatment. 3
Due to the rarity of BBS and its clinical manifestations, such as psychoses and schizophrenia, there is a paucity of literature on the subject. There are a few reports of psychosis in BBS in the literature (Table 1), but it is crucial to note that these were all cases of early-onset psychosis and only one case report mentioned management, 9 which is a shortcoming in the existing literature. Since late-onset psychosis has not been documented in BBS, our case is distinct.
Clinical characteristics and genetic abnormalities in individuals with psychiatric conditions associated with 15q11.2 deletion.
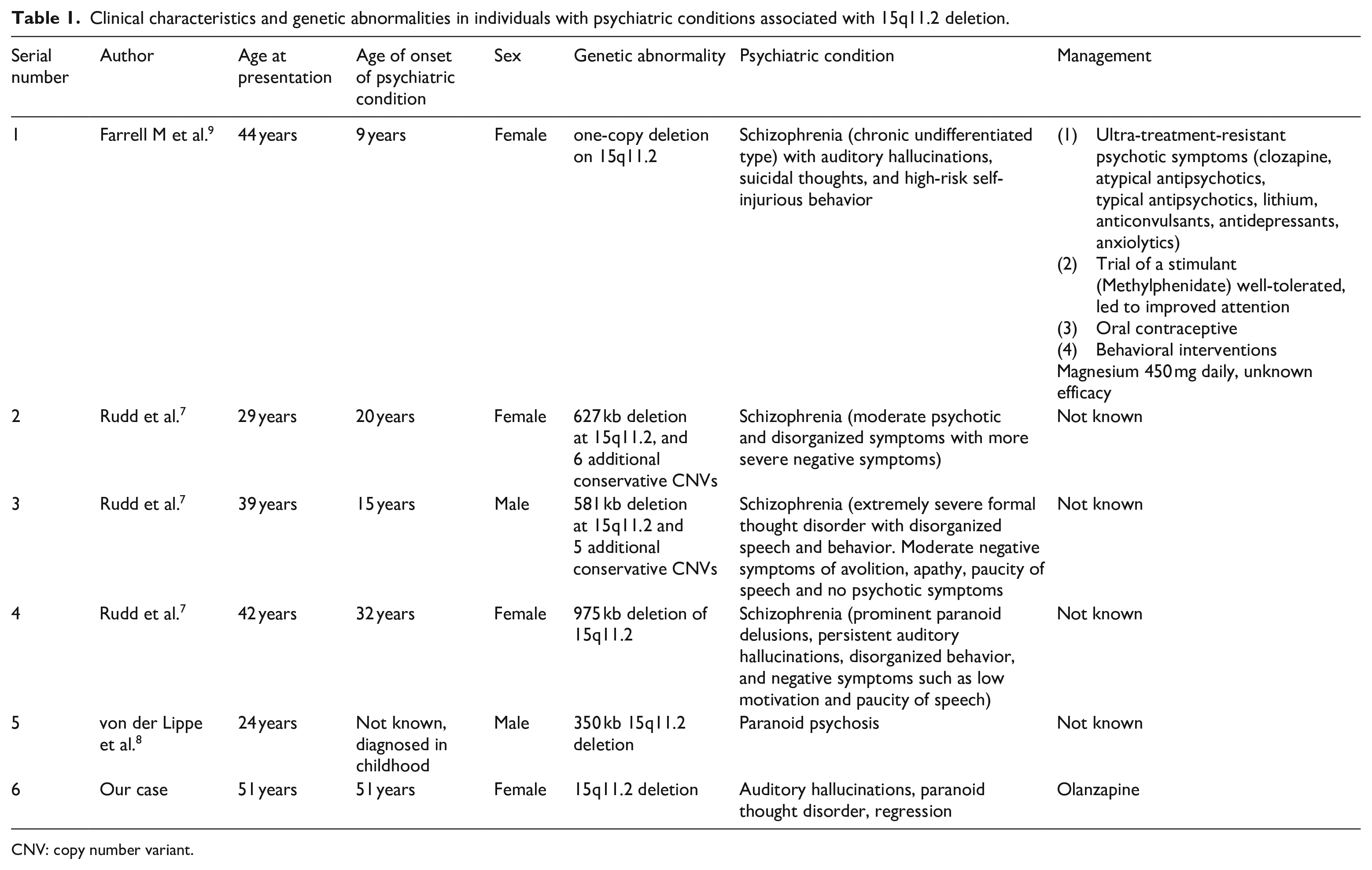
CNV: copy number variant.
Our case was that of a 51-year-old female patient with a history of mild intellectual disability, cerebral palsy, and a 15q11.2 BP1-BP2 microdeletion found during causal analysis of her cerebral palsy. She presented with auditory hallucinations and a decline in mental health. She had a long-standing dysthymia diagnosis and was treated with desvenlafaxine. No psychosocial stressors were reported when she presented with psychosis, ruling them out as triggers for an acute psychotic episode. Since her dysthymia was well managed with medication, progression to psychotic depression was ruled out. Concurrent intellectual disability and cerebral palsy greatly increase the risk of psychosis in adults,18–20 and increased age is associated with the occurrence of psychiatric disorders (including psychosis) in patients with cerebral palsy.21,22 This is a possible differential for our patient’s presentation.
The patient’s caregiver reported that the patient’s medical and psychiatric condition was at her usual baseline prior to the onset of this psychotic episode. The patient’s abrupt onset of psychosis could be a sign of neurodegeneration, making a dementing process a likely differential.23,24 This is a possible limitation in our case, as it is difficult to determine if the patient’s current presentation is due to microdeletion or neurodegeneration. Although a dementing process can be suspected as a cause for the patient’s symptoms, it is important to note that deletion carriers are not at risk for accelerated neurodegeneration compared to non-deletion carriers, as reported by Boen et al. 16
Despite being compliant with medications and stable for many years, she recently started exhibiting signs of a thought disorder. These signs included disorganized thought processes, paranoid beliefs, and delusions. Our patient specifically exhibited paranoid psychosis, which was also documented in another patient with early-onset psychosis in BBS. 8 Although she had thoughts of harming herself, she did not display the self-injurious behavior reported in BBS.1,9,20 The low phenotypic penetrance of BBS means that her psychotic symptoms may not have been directly caused by her BBS diagnosis. As the microdeletion has been classified as “pathogenic of mild effect size,” it is possible that these symptoms occurred coincidentally. The extent to which microdeletion is responsible for her symptoms remains unknown. Treatment with olanzapine proved effective in managing her symptoms.
This is in contrast to a case of BBS with early, childhood-onset, ultra-treatment-resistant psychosis reported in 2020 9 who showed no response to multiple antipsychotic medications, including clozapine. Despite polypharmacy, her clinical improvement was limited. A comprehensive evaluation was conducted, including neuroimaging, which showed no abnormalities except for an abnormal EEG (Electroencephalography). Her treatment was re-conceptualized, aiming to reduce unnecessary medications and implement behavioral interventions. Gradual tapering of staff support to prioritize independence and the introduction of methylphenidate and oral contraceptives led to improvements in attention, irritability, and anxiety. Knowledge of the 15q11.2 BP1-BP2 deletion had a positive impact on the treating team, facilitating a better understanding of the diagnosis and guiding treatment focus toward developmental delay, intellectual disability, and ADHD rather than solely psychosis. Magnesium supplementation was prescribed, although its efficacy was uncertain. The patient was reported to have made progress and was being planned for discharge.
Currently, comprehensive treatment guidelines lack addressing the psychiatric aspects of BBS. Treatment for individuals with BBS is personalized based on comprehensive evaluations such as neuropsychological, cognitive, and behavioral assessments by clinical geneticists, psychiatrists, psychologists, and developmental experts. Treatment also involves educational interventions, specialized therapies, and surgical or dental care for dysmorphic features. Behavioral and psychiatric disturbances require multidisciplinary healthcare involvement. Additionally, genetic counseling, prenatal detection, and regular assessments are essential. Referrals to specialized experts and early intervention programs need to be made accordingly.11,25 As a potential treatment option, Butler 12 suggested dietary magnesium supplementation since two of the four affected genes are involved in magnesium transport. However, it is imperative to note that this recommendation is supported by limited anecdotal evidence.
Conclusions
BBS displays varied clinical features. BBS individuals have developmental delays, speech issues, dysmorphic features, cognitive challenges, behavioral problems, and abnormal brain imaging.
Treatment guidelines for addressing psychiatric aspects of 15q11.2 BP1-BP2 deletion syndrome are lacking. Personalized approaches, including evaluations, multidisciplinary involvement, therapies, and assessments, are crucial. Genetic counseling, prenatal detection, and early interventions are vital.
Further research is necessary to understand the clinical manifestations and treatment of 15q11.2 BP1-BP2 microdeletion syndrome, particularly its psychiatric aspects.
Footnotes
Acknowledgements
The authors express their gratitude to the patient for her consent to publish this report.
Author contributions
S.D., M.R., and S.R. were responsible for conception, design, supervision, and partial drafting of the manuscript. V.S., S.P., and P.P. were responsible for data collection, analysis, and interpretation. They took part in literature review and drafting of the whole manuscript along with M.A. and O.Q., M.A., O.Q., M.R., and S.R. were also responsible for modifying the manuscript post peer review and provided critical review.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from the patient for her anonymized information to be published in this article. The patient regained fair insight and judgment over the course of her treatment regime to provide written informed consent by herself.