
Abstract
Being the second most common malignant bone tumor in children and young adults, Ewing’s sarcoma can also occur as a primary soft-tissue tumor called extraosseous or extra-skeletal Ewing’s sarcoma. It is a rare entity, especially in the pediatric population. We report the case of an adolescent who presented to our department for lower extremity magnetic resonance imaging to explore leg swelling. It revealed an extra-skeletal Ewing’s sarcoma with multiple bone metastases. By reporting this case, we also review the literature on this rare abnormality.
Introduction
In 1921, James Ewing described Ewing’s sarcoma (ES) for the first time as an osteolytic cancer of the bone composed of malignant small round cells. 1 It can rarely be of extra-skeletal origin (EES). These two entities belong to the ES family of tumors along with peripheral primitive neuroectodermal tumor (pPNET) and its subtype called the Askin’s tumor of the chest wall. 2 EES remains an enigmatic malignancy in the literature due to its lower frequency, later onset, and better prognosis compared to the classical ES. 3 Imaging modalities have an important role in diagnosing, staging, and in surveillance of these EES. 4 Magnetic resonance imaging (MRI) is the frequently preferred modality since it can assess the primary tumor and its local staging with high detection sensitivity for soft-tissue contrast. 5 We report the case of a 16-year-old boy who presented for an MRI with right leg swelling, which turned out to be an EES with multiple bone marrow metastases to both legs and thighs. Secondary pulmonary masses were later discovered.
Case
We report a case of a 16-year-old boy, with no significant medical nor past surgical history, who had been complaining of persistent pain in the right leg for a 2-month duration with the evolution marked by the extension of the pain to both thighs and knees and swelling in the right lower part.
The clinical examination noted tenderness in the right lower leg, irradiated to both thighs and knees and the estimated muscle tone was around 2/5 on the right foot. The patient was referred by the pediatrics department to our facility for an MRI of both lower limbs. MRI showed an ill-defined, lobulated soft-tissue mass of the inferior part of the right leg within the anterior tibial muscle, with an intermediate T2 signal containing some necrotic areas, a T1 heterogeneous low signal intensity (similar to skeletal muscle) with heterogenous but striking enhancement after Gadolinium administration (Figure 1). These findings were associated with multiple bilateral intramedullary osseous lesions of both the tibial and femoral bones. These lesions presented with an ill-defined limit, a heterogenous T2 hyper signal, and a T1 hypo signal heterogeneously enhanced after Gadolinium (Figure 2).

Coronal T1-WI and coronal and axial T1 with fat saturation after Gadolinium, showing the soft-tissue mass (arrow) within the tibialis anterior muscle in heterogenous hyposignal T1 (similar to skeletal muscle) with heterogenous striking enhancement after Gadolinium administration.

Coronal STIR (a and b), coronal (c) and axial T1-WI with fat saturation after Gadolinium (d and e), showing bilateral intramedullary osseous lesions of both the tibial (blue arrow) and femoral bones (red arrow), heterogeneously enhanced after Gadolinium.
These findings suggested a soft-tissue sarcoma with multiple bone marrow metastases. An excisional biopsy of the right soft-tissue mass of the lower part was performed to have histological confirmation. The latter showed marked tumor necrosis with high cellularity, composed of round uniform small cells, oval nuclei, and scanty lacy cytoplasm (Figure 3). There were some rare pseudorosettes (the equivalent of neuroectodermal differentiation) and immunopositivity for CD99 (diffuse-membranous) and Vimentin (Figure 4). A nonreactivity to cytokeratin, actin, neuron-specific enolase, and leucocyte common antigen was also noted. Confirmation of EES was established based on these histological and immunohistochemical findings. The patient then underwent a chest and abdomen computed tomography (CT)-scan which showed pulmonary nodules located in the right Fowler segment and the anteromedial segment of the inferior left lower lobe (Figure 5). The adolescent was then referred to the oncology department for chemotherapy and urgent medical care.

(a) Hematoxylin and eosin, 20x magnification. (b) Hematoxylin and eosin, 40x magnification. Histopathological examination of the soft-tissue mass showing sheets of monotonous small round blue cells associated to hyperchromatic nuclei and absence of nucleoli with some of these cells forming pseudorosettes.

Panels of immunohistochemical tests (40x magnification), showing the neoplastic cells diffusely positive for CD99 (a) and for vimentin (b).
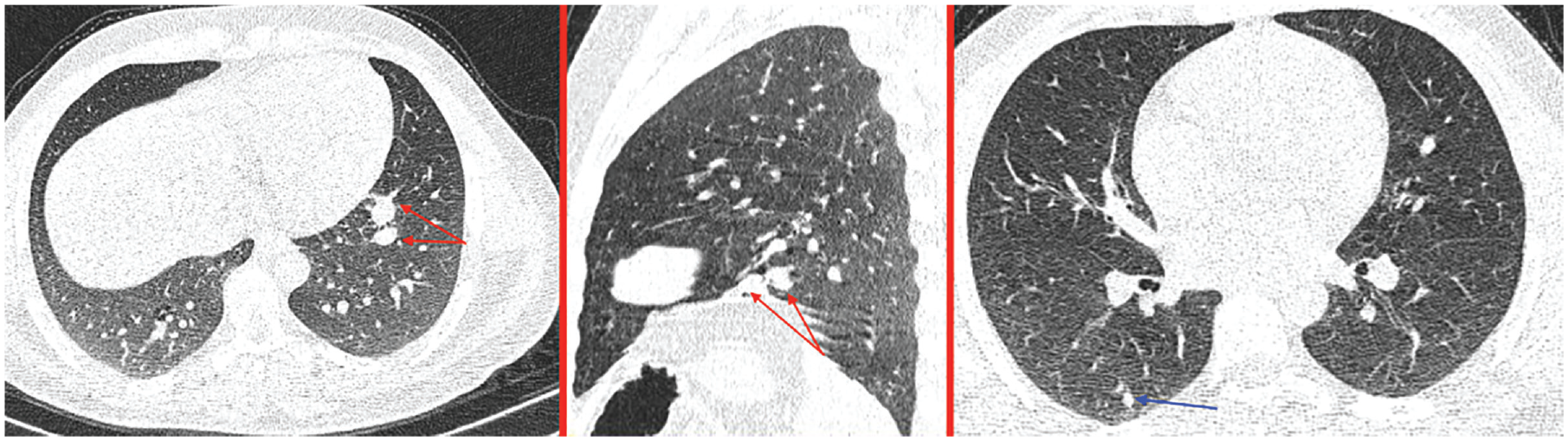
Axial chest CT (lung window) axial and sagittal reconstruction showing a nodule in the right Fowler (blue arrow) and two nodules of the antero-medial segment of the left lower lobe.
Discussion
ES is a childhood aggressive tumor, that may form in any part of the body and has a rapid growth tendency with a high rate of metastases. 6 It can rarely be of extra-skeletal origin, which was first described in 1969 as a malignant mesenchymal tumor (much rarer than the intraosseous entity). Its incidence is 0.4 per million, meaning it is 10 times less than that of ES. 7 As for its prevalence, it follows a bimodal distribution, peaking in those who are <5 years and >35 years. 3 This finding has been reported in previous studies and was consistent with a comparison of clinical features and outcomes in patients with extra-skeletal versus skeletal ES published in 2018 by Jiang et al. (an analysis of 3178 cases).8–10 This highlights the aim of our case report since it was found in an adolescent.
Other than age distribution, EES has other differences, compared to ES of bone such as no distinct male predilection, but instead a more equal distribution between sexes and the fact that the trunk is affected more frequently than the lower extremities. 11 Clinically, it may present as a superficial or deep soft-tissue mass, rapidly growing and usually solitary. Depending on its location, the tumor is generally a painless mass or may be associated with abdominal or chest pain. 12 In the paravertebral location, the mass may be present with cord compression symptoms. 13 Unfortunately, adult patients tend to have distant secondary lesions at presentation located in the lung, which is the most common site of metastasis. 12 Paravertebral regions and lower extremities are the most common location with approximately 30% and 25%. respectively. Chest wall, retroperitoneum, and upper extremities are also possible but are less common. An intracranial EES has also been noted, but it is extremely rare. 14
Imaging modalities will show a soft-tissue mass. On ultrasound, it will usually be a hypoechoic mass with an increased Doppler blood flow. On plain radiographs, we can have a normal appearance or a large soft-tissue mass with calcifications in 25% of cases. An adjacent bone erosion, cortical thickening, osseous invasion, or periosteal reaction may also be present but it is rare. 15 On CT scan, EES may present as a nonspecific soft-tissue mass of similar density to the muscle. A low attenuation can be present because of hemorrhage or necrosis and calcifications are present in 25% of cases. Similar to a plain radiograph, CT may show an osseous involvement if present. On MRI, we may evoke the diagnosis based on a common feature which is disorganized high-flow vascular channels associated with low signal intensity with all pulse sequences. A pseudo capsule may be seen with somewhat well-defined margins. On T1-WI, it is a heterogenous soft-tissue mass, prominently enhanced after Gadolinium administration but heterogeneously. On T2, it is a high or intermediate signal intensity with some fluid levels. Bone scintigraphy and fluorodeoxyglucose positron emission tomography may help show an increased radionuclide uptake. 16
Histopathology can confirm EES showing monotonous proliferation of small blue round cells solidly packed with intracellular glycogen which may indent nuclei. 17 Often, we can have necrotic regions demonstrating rich vascularity, and areas of hemorrhage. Almost always, there is a strong immunoreactivity for CD99 and vimentin. 18
We also note that staining is usually negative for leucocyte common antigen, CD30, Neuron-specific enolase, actin, and S-100, which enables us to exclude rhabdomyosarcoma as well as lymphoma. 19 To distinguish classic ES from pPNET, we have the absence of neurosecretory granules (neurofilaments). 19 Usually, morphology and immunohistochemistry are sufficient for the diagnosis. If there are unusual variants, molecular genetic analysis can confirm the EES, using fluorescence in-situ hybridization for EWSR1 rearrangement on formalin-fixed paraffin-embedded tissues. 20
In our case, immunohistochemistry revealed round cells with high cellularity, scanty cytoplasm, and positive reactivity for CD99 and vimentin.
Differential considerations of EES mainly include some higher grade vascular lesions such as haemangioendothelioma, haemangiopericytoma, and angiosarcoma with other malignancies in particular, rhabdomyosarcoma, synovial sarcoma and alveolar soft-part sarcoma.17,21 Neuroblastoma and lymphoma are less common considerations. 18 Rhabdomyosarcoma represents the most common soft-tissue malignancy in children. It is usually a painless tumor but rapidly growing. On MRI, it may have a T1 isointensity to muscle with some areas of hemorrhage and T2 hyperintensity with avid enhancement after Gadolinium administration. 21 As for non-rhabdomyo-sarcomatous childhood tumors, synovial sarcomas are the most common in lower extremities. It is a slow-growing mass with a preference for juxta-articular regions. 22 MRI findings are generally isointense signal to muscle mass on T1-WI, with heterogeneous hyperintense T2 signal due to necrosis, hemorrhage, and fibrosis bands. This combination forms a characteristic multilobulated lesion separated by septa, which forms large cystic foci with regions of hemorrhage giving it “bowl of grapes appearance.”22,23
Recent studies have identified smaller groups of sarcomas that display a limited overlap with ES and regrouped them in an informal label of Ewing-like sarcoma. As a result of a unique combination of gene fusion between EWSR1/FUS and the genes encoding the erythroblast transformation specific (ETS) family of transcription factors, ES was reclassified in the most recent World Health Organization classification of soft-tissue and bone malignancies. 24
Additionally, three distinct groups of Ewing-like sarcomas were recognized by the classification: CIC-rearranged sarcoma, sarcoma with BCOR genetic alterations, and round cell sarcoma with EWSR1::non-ETS fusions. However, it is likely that the last group does not represent a single entity because it includes tumors with a variety of fusions and phenotypes. 25
Managing the disease should start with neoadjuvant chemotherapy which represents the standard of care. Then comes a definitive radiation or surgery. 26 EES has a high propensity for local recurrence and distant metastasis, which was the case of our patient who had multiple bone and lung metastasis. His prognosis is unfortunately poor.
Conclusion
Although considered a rare tumor in the pediatric population, EES should be evoked in young adults presenting with a circumscribed, large, and heterogenous mass in the lower extremities or paravertebral regions. MRI can have an impact on narrowing the differential diagnosis and also in assessing the tumor’s local staging. Other imaging modalities help in evaluating lung and other distant or nodal metastases. Nowadays, immunohistochemical examination and genetic study can confirm the diagnosis.
Footnotes
Author contributions
Y.E.H., was responsible for article concept, design as well as editing, and literature search; S.C., helped in editing and reviewing of the article and literature search; K.L., contributed to the conception and design; N.A., contributed to acquisition, analysis, and interpretation; L.C., critically revised the article and gave final approval; S.E.H., contributed to the acquisition, analysis, and interpretation, critically revised the article, and gave final approval.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.