
Abstract
Trichohepatoenteric syndrome or syndromic diarrhea is a rare and severe Mendelian autosomal recessive syndrome characterized by intractable diarrhea, facial and hair abnormalities, liver dysfunction, immunodeficiency and failure to thrive. It has been associated with mutations in TTC37 and SKIV2L genes, which encode proteins of the SKI complex that contributes to the cytosolic degradation of the messenger RNA by the cell’s exosome. We report a case of a male infant who suffered from typical symptoms and signs of trichohepatoenteric syndrome without immunodeficiency. The patient’s genetic testing showed a very rare mutation in SKIV2L gene’s 25 exons (p.Glu1038 fs*7 (c.3112_3140del)). Even though our patient was provided with total parenteral nutrition from birth, the child’s death in the third year of age highlights the severity of the disease and the poor prognosis of this particular type of genetic predisposition.
Keywords
Introduction
Trichohepatoenteric syndrome (THE) or syndromic diarrhea (SD) was first described in 1982 by Stankler et al. and furthermore in 1994 by D Girault et al. as a clinical entity characterized by severe infant diarrhea combined with physical abnormalities and deficiencies of the immune system.1,2 Until nowadays, there are no sufficient epidemiological data for the disease. In Western Europe, there is an estimated prevalence of 1/300,000–1/400,000 live births. 3
Mutations in one of two specific genes have been reported in the recent bibliography to cause the syndrome with a Mendelian autosomal recessive pattern of transmission. These genes encode the tetratricopeptide repeat domain–containing protein 37 (TTC37) and the superkiller viralicidic activity 2 (SKIV2L). These are proteins of the SKI complex, which is a co-factor of the RNA exosome in the cytoplasm.4–7 Exosomes are well preserved in all eukaryotic cells. As a result, yeast helped to identify the role of the superkiller complex (SKI complex), which is to contribute as a co-factor in the messenger RNA (mRNA) cytoplasmic degradation of the RNA exosome. The TTC37 gene encodes the tetratricopeptide repeat protein 37 (Ski3 in yeast) and the SKIV2L encodes the helicase SKI2W (Ski2 in yeast).4,5
The phenotype of THE/SD is characterized by a wide variety of symptoms, signs and physical abnormalities. The most common of them are intractable diarrhea, hair abnormalities and facial abnormalities, such as prominent forehead and cheeks, broad nasal root and hypertelorism. Other signs and symptoms include immunodeficiency, low birth weight and failure to thrive.8,9 Hepatic function is commonly affected while skin abnormalities (in most cases café au lait spots) are not rare. Congenital heart defects and platelet anomaly have a lower frequency of incidence. 9
Regarding the management of the disease, a lot of effort has to be done in order to establish a treatment schedule due to its rarity, which results in a lack of evidence-based treatments. The treatment of each young patient still relies on the clinician’s experience and knowledge in each center. The main treatment consists of the parenteral nutrition (PN) and supportive therapy in each case according to its needs. 10 In patients with severe hepatic involvement, total parenteral nutrition (TPN) is contraindicated. 11 As a result, the survival of the patients varies significantly between different cases. 12
Case report
The reported case is a male infant of Roma ethnicity who suffered from intractable diarrhea and failure to thrive. During his mother’s pregnancy, he was diagnosed with intrauterine growth retardation (IUGR). He was born small for gestational age (SGA)—39 weeks, weighing 2.45 kg (<3rd percentile) with a head diameter of 33.5 cm (10th percentile) and height of 44.0 cm (<3rd percentile). He was the third child of the mother (two of the patient’s siblings had a different father). After the labor, he was immediately taken to the emergency department diagnosed with sepsis and neonatal respiratory distress syndrome.
After the septic episode had been managed in the neonatal intensive care unit, the infant was admitted, at the age of 2.5 months, to our pediatric department. The reasons for the admission were intractable diarrhea (four to five episodes per day of watery stools mixed with mucous secretions but without macroscopically blood loss) and failure to thrive. Patient’s IUGR, intractable diarrhea from the first day of life and failure to thrive prompted a wide range of diagnostic investigation first in our department and subsequently at the Children’s Hospital in Athens where the infant was referred and remained hospitalized in order to complete the diagnostic evaluation. The investigation processes included cystic fibrosis, microvillus inclusion disease, immunodeficiencies (white blood cell (WBC) count and type of cells, levels of serum immunoglobulins, subclasses of IgG, phagocytic sufficiency of macrophages), autoimmune enteropathy, tufting enteropathy, disaccharidase deficiencies, glucose-galactose malabsorption and food allergies, without success in the establishment of a diagnosis. Upper gastrointestinal (GI) endoscopy and colonoscopy revealed mild villous atrophy and some eosinophilic infiltration of the short bowel mucosa. On biochemical investigation, elevated liver enzymes (about two times over the upper normal limit, alanine aminotransferase (ALT): 121 mg/dL, aspartate aminotransferase (AST): 113 mg/dL were a standard finding, while the immunophenotype, the karyotype and the serum and urine amino acid levels were normal.
At the age of 6 months, the infant was readmitted at our pediatric department and remained hospitalized due to persistent diarrhea, failure to thrive (body weight and body length <3rd percentile, 5.5 kg and 60 cm, respectively) and the need for nutritional support. The infant’s clinical characteristics, which were prominent forehead and cheeks, broad nasal root, widely spaced eyes (hypertelorism), wooly hair that was easily removed and poorly pigmented at the age of 11 months, are shown in Figure 1. Further investigation showed a mild congenital heart atrial septal defect, while hospital discharge was not feasible because of an unsafe living environment due to the low socioeconomic and educational status of the family and parents’ feeling of insecurity.

The infant at the age of 11 months.
The combination of signs and symptoms consisting of intractable diarrhea present from birth, failure to thrive, through enteral nutrition, persistent facial and hair dysmorphic features (Figure 2) and elevated liver enzymes led to genetic testing (targeted Sanger sequencing—Sequencage direct de la region codante du gene SKIV2L [direct sequencing of the gene coding region SKIV2L]) which was positive for homozygous deletion of 29 nucleotides, p.Glu1038 fs*7 (c.3112_3140del), in exon 25 of the SKIV2L gene leading to a frameshift of the open reading frame (ORF) while being negative for a potential TTC37 mutation. The infant was finally diagnosed with the THE as he was homozygous for an autosomal recessive mutation of the SKIV2L gene (MutationTaster—Figure 3). The diagnosis was made at the age of 2 years.
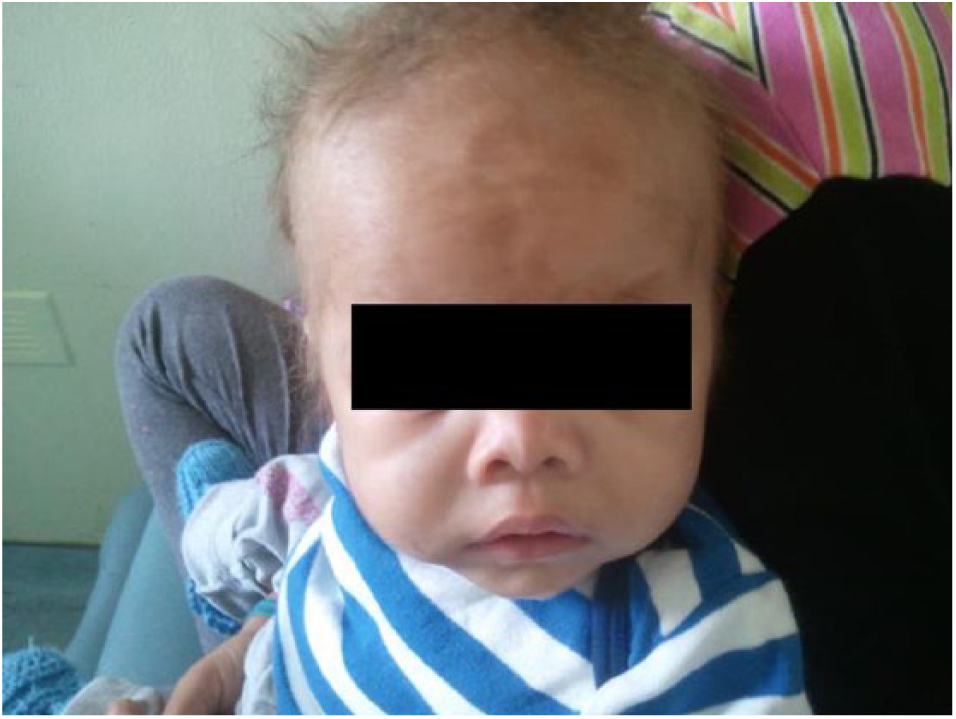
The infant at the age of 15 months.

MutationTaster.
The patient was on TPN from birth due to severe diarrhea and inability to thrive. The use of an enteric elemental formula failed to improve diarrhea and body weight. The results with TPN were more promising in regard to weight gain (Figure 4). In a period of about 2.5 years of hospitalization and continuous TPN, the child experienced three episodes of septicemia, one episode of fungemia, three episodes of hypoglycemia and thrombosis of the three central veins, that is, the two subclavian veins and the femoral vein. TPN from birth combined with hepatic involvement due to the syndrome led to complete hepatic failure, portal hypertension and GI bleeding because of which the child was admitted to the pediatric intensive care unit. Α fourth episode of septicemia while being treated in the intensive care unit led to multiple organ failure and finally death.
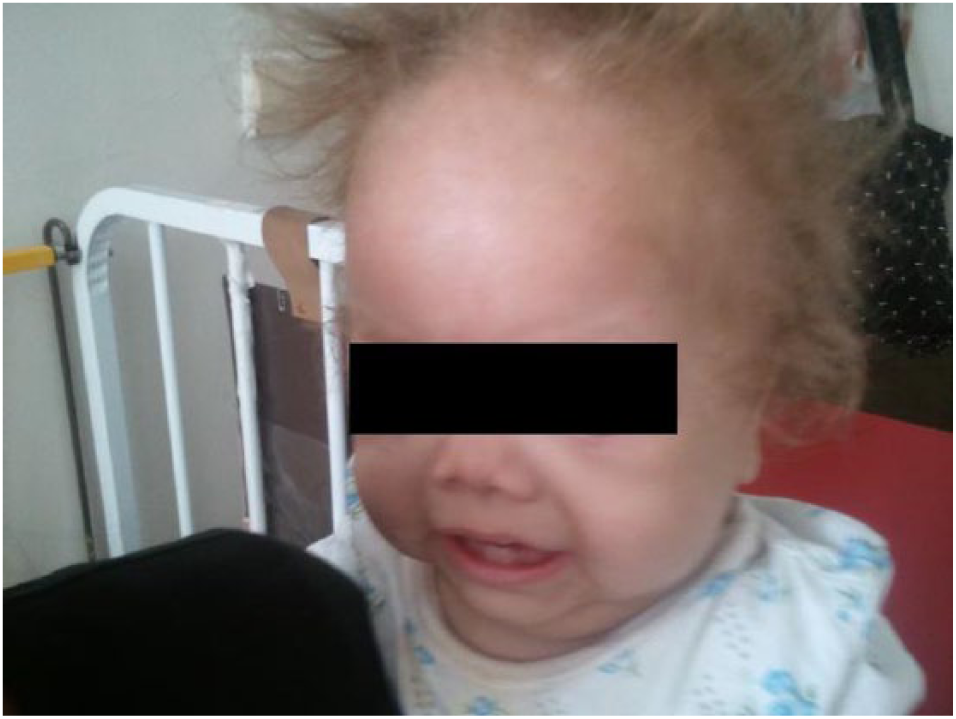
The infant at the age of 21 months.
Discussion
THE should always be included in the differential diagnosis of an infant with persistent diarrhea between birth and failure to thrive. If the aforementioned symptoms are associated with hair and facial abnormalities with or without immunodeficiency, a genetic test is of great importance. Although being a rare clinical entity, THE awareness is increased among physicians in recent years, as there is a need for specific criteria for establishing the diagnosis. Up to now, cases have been described in Indian, European and Mediterranean populations. 9
Most of the THE cases are TPN dependent and just a small percentage achieve normal oral nutrition after many years. Many of these cases have a fatal outcome due to TPN complications. Small bowel transplantation is not a treatment of choice. Combined liver and small bowel transplantation represent an even more complicated and dangerous approach. Long-term PN is the only currently available treatment.9,10 Furthermore, there is no previously reported case, to our knowledge, of a child with THE who was subjected to liver transplantation alone or to a combined liver and small bowel transplantation.
Fabre et al. reported 48 cases who all had intractable diarrhea from infancy, defined as chronic diarrhea, persistent despite an enteral rest by means of TPN. The onset of diarrhea varied from the first day of life up to 32 weeks after birth. 9 Moreover, they described hair abnormalities in all cases. Facial dysmorphism was present in 47 out of 48 cases. Children presented with a wide forehead, broad nasal root, hypertelorism, coarse features and immune defects in 39 out of 44 cases. IUGR combined with SGA was present in 31 out of 46 patients, while the presence of IUGR/SGA combined with liver involvement was present in 23 out of 44 patients (23/44). More than half of the children had liver disease with cirrhosis and siderosis being the cardinal features. Hepatomegaly was a rare feature, while hepatoblastoma was described in one patient. Cardiac abnormalities (aortic insufficiency, peripheral pulmonary stenosis, atrial or ventricular septal defect) were found in 8 out of 31 children. Hartley et al. reported an increased platelet size found in 5 out of 21 patients, which was temporary in some cases. 9
In 2014, in a French cohort study with 15 THE cases, the authors report that all patients presented typical SD/THE syndromic features, such as intractable diarrhea in infancy requiring PN, a facial dysmorphism with hair abnormalities and immunological disorders. Half of them also had liver and skin abnormalities. Five children died, of which three died due to infections. Probabilities of survival according to the Kaplan–Meier method were 93.3%, 86.7%, 74.3%, and 61.9%, at 1 year, 5, 10 and 15 years of age, respectively. Three patients weaned from PN with a likelihood of weaning being 10% at 5 years and 40% at 10 years. At birth, 80% were SGA, while the short stature persisted in 60%. Hemophagocytic syndrome was noted in 60% of the cases, while a mild mental retardation was present in 60% of the cases. 8
In another more recent report, the authors reviewed the literature conducted in May 2017 and found 29 articles and two abstracts that were included describing a total of 80 patients, of which 40 presented with mutations in the TTC37 and 14 cases with mutations of the SKIV2L genes. 10 Moreover, they found that PN was used in the management of 83% of the patients while weaning from PN was possible in 44% of the cases (mean duration = 14.97 months). Immunoglobulins were used in 33 patients, but data on efficacy were reported in 6 patients with a diminution of infection (n = 3) or diarrhea reduction (n = 2). One-third of the patients (24/80) died at a mean age of 23.5 months, mostly, from infection (7/24) or hepatic failure (7 patients) for the 18 patients with recorded information. The authors finally concluded that the management of SD/THE mainly involves PN and immunoglobulin supplementation. Antibiotics, steroids, immunosuppressants and hematopoietic stem cell transplantation are not recommended as principal treatments since there is no evidence of efficacy.
Our patient was diagnosed with IUGR and was an SGA neonate. He manifested intractable diarrhea and failure to thrive since birth and was TPN dependent. Moreover, he manifested all the clinical features of THE syndrome which were prominent forehead and cheeks, broad nasal root, widely spaced eyes (hypertelorism), wooly hair and had heart involvement (atrial septal defect). No immunodeficiency was detected. Histological assay of the short bowel mucosa revealed mild villous atrophy. Moreover, hepatic involvement led progressively to hepatic failure.
Therapeutic approach was with nutritional support by means of TPN from which he never weaned. In addition, he suffered from recurrent systematic infections due to central vein catheters. Death was due to sepsis and hepatic failure. According to the above-mentioned literature, our case was in accordance with previously reported cases and had a progressive deterioration of his health status with a fatal outcome in the third year of life.
The mutation observed in our case (homozygous deletion of 29 nucleotides (c.3112_3140del) in exon 25 of the SKIV2L gene) is the second one to be reported globally. The first was reported in 2018 from Bourgeois et al. Moreover, our patient is the first reported THE case in Balkan. The fact that our patient did not suffer from immunodeficiency may be due to the different exon affected, indicating that there may be different phenotypes of this genetic defect and that this specific defect in our patient may have a more severe clinical course and outcome.
Recently, the first Japanese THE female patient was described with causative novel compound heterozygous mutations in the SKIV2L gene.
Sequencing analysis showed compound heterozygous nonsense mutations, c.1420G>T (p.Q474*) of the paternal allele and c.3262G>T (p.E1088*) of the maternal allele, in the SKIV2L gene. No mutation was found in TTC37. Flow cytometry indicated a decreased expression of the SKIV2L protein in peripheral blood mononuclear cells (PBMCs) from the patient compared with that in a healthy control sample. 13 The patient was put on TPN for 30–100 days and after many years, at the age of 13 years, she started a normal diet. 13
Others in 2018 identified an autosomal recessive C.1891G>A missense mutation (NM_006929) in SKIV2L gene that was previously described only in a compound heterozygous state as causing THE syndrome in a child with failure to thrive and chronic diarrhea but without hair or facial abnormalities. 14 The authors report that protein modeling suggested that the mutation has the potential to cause structural destabilization of SKIV2L, through conformational changes, interference with the protein’s packing or changes at the protein’s interface.
At the same time, Fabre et al. presented a detailed description of seven patients, of two families, with the same novel mutation in TTC37: c.4572 G>A (p.Trp1524X). All patients were homozygous for this mutation and presented the typical clinical features of SD/THE, but with a milder presentation than usual. 15
In a recent study, the authors report the phenotype and genotype analysis of a cohort together with an extensive literature review of THE cases worldwide, that is, 96 individuals harboring mutations in one gene or the other. They set up locus-specific databases for both genes and reviewed the type of mutation as well as their localization in the proteins. They reported that no hotspot is evidenced by any type of mutation. The phenotypic analysis was first made in the whole cohort but was limited due to heterogeneity in clinical descriptions. Then, they examined the lab diagnostic cohort in detail for clinical manifestations. They stated that “for the first time, we are able to suggest that patients lacking SKIV2L seem more severely affected than those lacking TTC37, in terms of liver damage and prenatal growth impairment.” 16
This finding is in accordance with our case in which the SKIV2L mutation caused a more severe clinical phenotype with a fatal outcome.
More recently, Vély et al. 17 reported nine cases with THE (three with SKIV2L and six with TTC37 mutations). They searched more specifically for various immunodeficiencies and found several immunological defects such as very-low-switched memory B lymphocyte count, impaired IFN-γ production by T and natural killer (NK) cells and reduced degranulation of NK cells. Moreover, T-cell proliferation was abnormal in three of the six TTC37-mutated patients.
In our patient, a simple search for immunodeficiencies (classes and subclasses, phagocytic ability, WBC and type) did not reveal an immune system instability. After the diagnosis was made, we planned for more specific evaluation of the immune system of the baby but rapid worsening of the health status of the baby did not allow this option.
In conclusion, further experience and knowledge are needed regarding the management of this rare syndrome since various genetic mutations could play a role in different treatment options and outcomes. Many more cases need to be reported in order to gain knowledge in regard to the diagnosis, management, and prognosis of this severe and usually fatal disease.
Footnotes
Acknowledgements
We would like to thank cordially the colleagues of the Aglaia Kyriakou Children’s Hospital in Athens and especially Dr Alexandra Papadopoulou who accepted the child at the Department of Gastroenterology and completed the diagnostic workup in terms of endoscopic evaluation and the histological and biochemical investigation of the obtained specimens due to the advanced technical equipment of the department. Our best thanks to colleague Alexander Fabre who helped us to make possible the genetic testing in the Laboratoire de Génétique Moléculaire, Hôpital d’enfants de la Timone, Marseille. Best thanks to biologist Dr Efstratios Kosmidis for the support in order to give our best answers to the reviewers’ comments in the field of biology.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article and consent was obtained to publish the subject images.