
Abstract
Wiskott–Aldrich syndrome is a rare X-linked recessive disease resulting from variations in the WAS gene. Wiskott–Aldrich syndrome is sometimes difficult to differentiate from immune thrombocytopenic purpura. A 2-month-old boy was admitted to our hospital for purpura and thrombocytopenia. His mean platelet volume was reported to be normal. Treatment with intravenous immunoglobulins failed to improve the patient’s platelet count. Subsequently, an acute cytomegalovirus infection was confirmed by serological testing and antigenemia. The patient was diagnosed with immune thrombocytopenic purpura secondary to a cytomegalovirus infection. However, based on the patient’s clinical course and the refractoriness of his condition, Wiskott–Aldrich syndrome was strongly suspected. Through direct sequencing of the genomic DNA of the Wiskott–Aldrich syndrome protein (WASP) gene, we identified a novel missense mutation in exon 3 of the patient’s WASP gene (c. 343 C>T, p. H115T), and the patient was diagnosed with Wiskott–Aldrich syndrome at 3 months after onset. Children with Wiskott–Aldrich syndrome are often initially diagnosed with immune thrombocytopenic purpura, which can lead to inappropriate treatment and delays to life-saving definitive therapy. Our findings imply that Wiskott–Aldrich syndrome should be considered as a differential diagnosis in cases of refractory immune thrombocytopenic purpura combined with a cytomegalovirus infection.
Introduction
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disease resulting from variants in the WAS gene. It is characterized by a triad of immunodeficiency, eczema, and thrombocytopenia. WAS is sometimes difficult to differentiate from immune thrombocytopenic purpura (ITP). 1 We describe the case of a 2-month-old boy with WAS who was initially diagnosed with ITP secondary to a human cytomegalovirus (HCMV) infection.
Case
A 2-month-old boy was admitted to our hospital with purpura, bloody stools, and thrombocytopenia. His medical history included a recurrent skin rash, which first presented at birth. On admission, he was generally well, but had purpura and hepatosplenomegaly (liver: 2 cm below the right costal margin and spleen: 5 cm below the left costal margin). His platelet (PLT) count was 10 × 109 L−1, his hemoglobin level was 7.6 g/dL, and his leukocyte count was 15.9 × 109 L−1. His mean platelet volume (MPV) was 9.9 fL (normal range: 8.9–12.6 fL). Although a bone marrow examination revealed normal cellularity without any malignant cells, it did not reveal hypermegakaryocytes. Subsequently, an infectious laboratory workup was performed. An HCMV infection was confirmed because positivity for the specific antibody against HCMV immunoglobulin M and HCMV antigenemia (23 of 48,000 cells were positive) were detected. There was no laboratory evidence of a maternal cytomegalovirus (CMV) infection during pregnancy. Based on these findings, the patient was diagnosed with ITP secondary to a CMV infection acquired after birth. An ophthalmic examination and audiogram produced normal findings. He received an intravenous infusion of immunoglobulins (IVIG: 1 g/kg/day) as a first-line therapy. Although the patient’s PLT count temporarily rose (118 × 109 L−1) following the IVIG therapy, it gradually decreased to 10 × 109 L−1 within a week. Treatment with prednisolone (2 mg/kg/day) failed to improve the patient’s PLT count, and repeated PLT transfusions were required. The intravenous administration of ganciclovir (10 mg/kg/day) was initiated for the CMV infection. Although no CMV antigenemia was present after 1 month, CMV antigenemia was seen again after the cessation of ganciclovir. Based on the patient’s clinical course and the refractoriness of his condition to therapies targeting ITP and CMV antigenemia, WAS was strongly suspected. Flow cytometric (FCM) analysis revealed that the level of WAS protein (WASP) expression on the CD3-, CD19-, and CD56-positive cells from the patient was only 20% of that seen in the control (his father). Through direct DNA sequencing, we identified a novel missense mutation in exon 3 of the patient’s WASP gene (c. 343 C > T, p. H115T; Figure 1). This mutation has not been described previously. The patient was diagnosed with WAS at 3 months after the onset of his condition. Subsequently, he underwent a cord blood transplant from an unrelated donor after receiving a conditioning regimen consisting of busulfan and cyclophosphamide. Ganciclovir was continued during the hematopoietic stem cell transplantation (HSCT) until CMV antigenemia was no longer detected (Day 180). At present, the patient is 9 years old and well. His PLT count is within the normal range, and 100% donor engraftment was achieved.
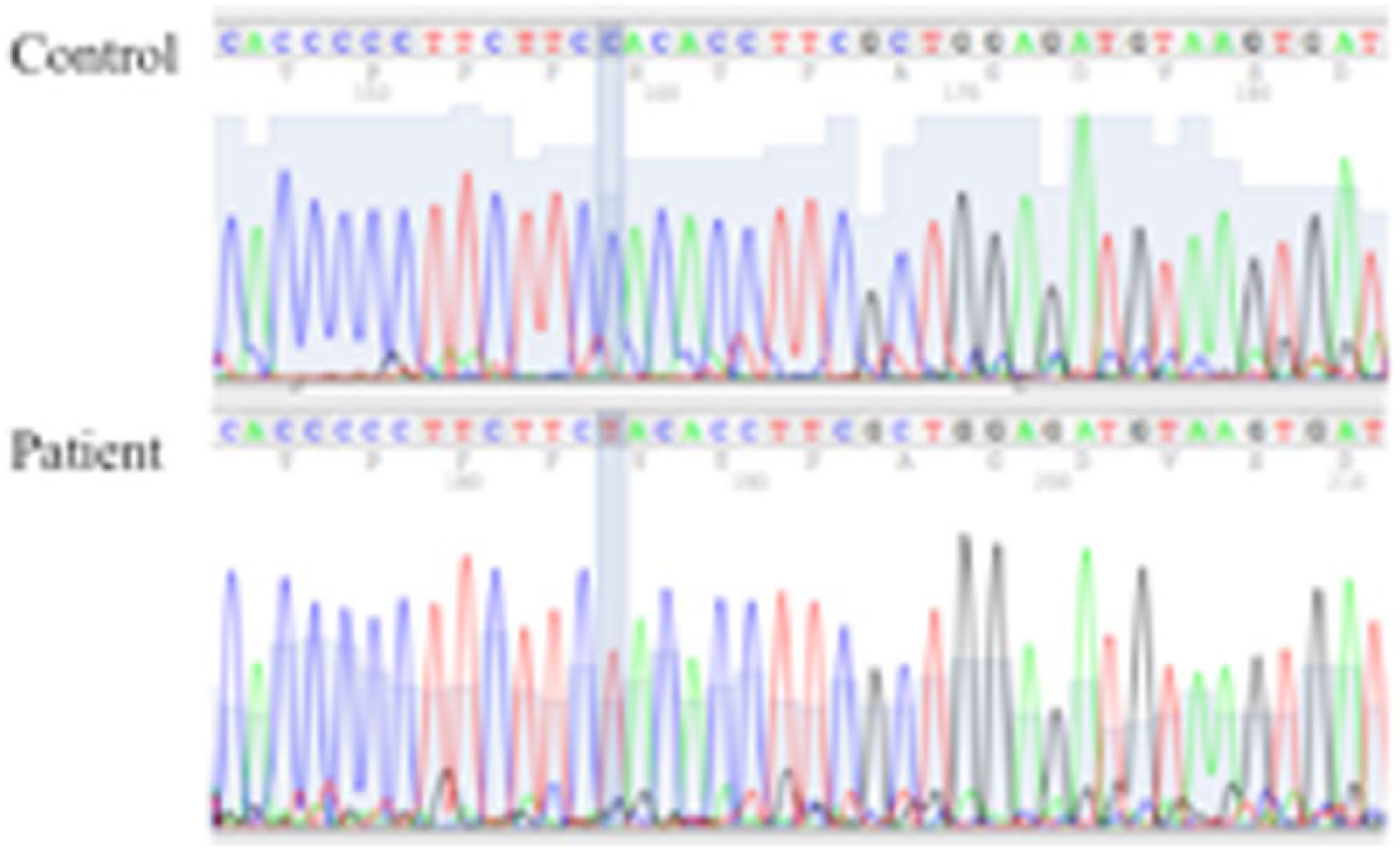
Chromatogram showing the DNA sequences of a healthy normal control (the patient’s father) and the patient. The patient had a C > T mutation, which resulted in the 115th amino acid changing from histidine to threonine.
Discussion
Children with WAS are often first diagnosed with ITP, potentially leading to both inappropriate treatment and a delay in definitive life-saving therapy. In addition, WAS is ultimately diagnosed in 7% of cases of conditions that are often mistaken for ITP. 2 In this case, the typical symptoms of WAS (thrombocytopenia, bloody stools, and eczema) were present on admission. However, a diagnosis of WAS was not considered because of three unusual features. First, the patient’s PLT size was normal, and he had thrombocytopenia. WAS is traditionally differentiated from ITP based on the small size of the PLTs seen in WAS patients. 1 However, the clinical phenotype of WAS itself is very heterogeneous, and some WAS patients have been found to have normal MPV.1,3 The clinical phenotypes of WAS, including PLT size, depend on the mutations present in the WAS gene. 3 In this case, a new mutation (c. 343 C > T, p. H115T) was identified, and it might have been responsible for the patient’s normal PLT size. Second, the initial IVIG therapy resulted in a rapid increase in the patient’s PLT count. Although some WAS patients who received IVIG therapy exhibited increases in their PLT counts, it is considered that IVIG therapy does not affect the PLT counts of WAS patients. 4 The reaction to IVIG therapy was temporary in our case. Therefore, IVIG treatment ultimately did not improve the patient’s PLT count. Finally, our patient had a CMV infection concomitant with thrombocytopenia. CMV is one of the most common causes of thrombocytopenia, including ITP. 5 Therefore, we consider that our patient’s thrombocytopenia was caused by his CMV infection, and so he was diagnosed with ITP secondary to a CMV infection. However, WAS patients are easily infected with CMV, and we should consider the presence of WAS as a basic disease. In conclusion, cases of persistent thrombocytopenia (regardless of the patient’s PLT size), particularly those involving concomitant CMV infections, should raise a suspicion of WAS. We recommend that a combination of FCM-based WASP screening and genetic studies should be performed in such cases.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.