
Abstract
Background:
We report a novel case of a rare disease: spontaneous Creutzfeldt–Jakob disease in a patient with well-controlled HIV. We explore the relationship between spontaneous Creutzfeldt–Jakob disease and HIV.
Case report:
A 66-year-old man with long-standing, well-controlled HIV infection presented with 3 months of progressive, subacute neurocognitive decline. His symptoms included conceptual apraxia, apathy, memory impairment, and gait disturbance, and were initially attributed to depressive “pseudo-dementia.” Unfortunately, the patient’s symptoms rapidly progressed and he ultimately succumbed to his illness. Autopsy confirmed the clinical diagnosis of spontaneous Creutzfeldt–Jakob disease.
Discussion:
This case highlights spontaneous Creutzfeldt–Jakob disease as a rare terminal illness in the setting of well-controlled chronic HIV. To our knowledge, this is the first report of a patient with chronic and previously well-controlled HIV infection dying from a prion disease. Despite the very different epidemiology and pathophysiology of HIV and spontaneous Creutzfeldt–Jakob disease, this case does raise questions of whether certain host genetic factors could predispose to both conditions, albeit currently, there is no clear causal link between HIV and spontaneous Creutzfeldt–Jakob disease.
Keywords
Introduction
Diseases of the central nervous system (CNS) caused by prions include Creutzfeldt–Jakob disease (CJD) and CJD-variant, Kuru, Gerstmann–Sträussler–Scheinker syndrome, and fatal familial insomnia.1–4 Prion diseases are neurodegenerative disorders with variable incubation periods, but are inexorably progressive and fatal once clinical symptoms appear. 1 The term “prion” was first coined by Dr Stanley Prusiner in 1982 to signify a small infectious pathogen containing protein but apparently lacking nucleic acid. 4 Histopathological findings include neuronal loss, gliosis, and spongiform vacuolization of the neuropil in the absence of a neuroinflammatory response.2,5 One of the characteristics of prion disease is their resistance to normal decontaminating procedures. 6 In humans, the gene encoding PrP is located in the short arm of chromosome 20, 7 and different mutations in the PrP gene have been linked to familial prion diseases.7–10 CJD is the most frequent of the human prion diseases, although still rare, occurring at a frequency of approximately one case per million worldwide in its sporadic form. 11 Familial CJD is thought to contribute to less than 15% of all CJD cases, 11 and iatrogenic CJD is increasingly less prevalent.
In this case report, we describe a novel case of a rare disease: spontaneous Creutzfeldt–Jakob disease in the setting of well-controlled HIV. To our knowledge, this is the first report of a patient with chronic and previously well-controlled HIV infection succumbing to a prion disease. We raise questions on whether certain host genetic factors could predispose to both conditions.
Case report
A 66-year-old man with long-standing, well-controlled HIV infection, Hepatitis B, hypertension, and peripheral vascular disease presented with 3 months of progressive, subacute neurocognitive decline. His symptoms included conceptual apraxia, apathy, memory impairment, and gait disturbance, which were initially attributed to “pseudo-dementia,” as well as ataxia with gait disturbance which was also attributed to polypharmacy, along with chronic peripheral neuropathy. Despite adjusting his medication regimen, his neurocognitive symptoms progressively worsened, and in the week preceding his transfer to our institution, culminated in an admission to an outside hospital for altered mental status and aspiration pneumonia.
On admission to our institution, the patient was cachectic and chronically ill appearing. His temperature was 101.2°F, blood pressure 140/82, heart rate 90 bpm, respiratory rate 20 breaths/min, pulse oximetry 99% on 2 L/min supplemental oxygen, weight 137 lbs, and height 5′ 10″. His physical examination was notable for diffuse muscle wasting, mildly increased work of breathing, and diffuse rhonchi on auscultation. He was minimally interactive and mute, but followed some basic commands. His cranial nerve examination showed no focal deficits. Motor examination showed increased tone in all extremities with atrophic bulk and bilateral upper extremity clonus with forearm flexion. There was marked bilateral upper extremity hyperreflexia with 3+ deep tendon reflexes at biceps and brachioradialis, though no reflexes were elicited in the lower extremities.
Initial laboratory work-up included a complete blood count which was notable for new pancytopenia, with a white blood cell count of 1.4 (×109 L−1), hemoglobin 7.9 g/dL, and platelet count 64,000 mL−1. Initial pancytopenia was thought to be related to sepsis of pulmonary origin. Chemistries were notable for Na of 152 and Cl of 122 which corrected with free water administration without change in the patient’s mentation. CD4 count 1 month prior was 531 µL−1 and HIV-1 RNA was undetectable. Lumbar puncture was obtained and showed a cerebrospinal fluid (CSF) opening pressure of 16 cm H2O, total protein 59 mg/dL (range, 15–50 mg/dL), glucose 56 mg/dL (serum glucose, 114 mg/dL), red blood cells (RBCs), and 0 nucleated cells. Additional CSF studies were performed including Epstein–Barr virus (EBV), herpes simplex virus (HSV), and cytomegalovirus (CMV) virus polymerase chain reaction (PCR), venereal disease research laboratory (VDRL), cryptococcal antigen, Histoplasma antigen, bacterial and fungal culture, and John Cunningham (JC) virus PCR which were all negative. Serum cryptococcal antigen and Toxoplasma IgG were also negative, as was nasopharyngeal swab PCR for influenza A/B, respiratory syncytial virus, and rhinovirus. CSF 14-3-3 protein returned positive, while T-Tau protein was 11,406 pg/mL (normal range, 0–1149 pg/mL), and real-time quaking-induced conversion (RT-QuIC) analysis was positive. Those three markers were interpreted by National Prion Disease Pathology Surveillance Center (Cleveland, OH) as suggesting ⩾98% probability of prion disease.
Magnetic resonance imaging (MRI) of the brain with and without gadolinium contrast was obtained at the outside hospital prior to transfer and was notable for prominent diffusion restriction along the bilateral caudate, putamen, and thalami, as well as gyriform cortical restricted diffusion in the bilateral medial cerebral hemispheres (Figure 1). Continuous electroencephalogram (EEG) monitoring was obtained and demonstrated diffuse slowing, without evidence of seizure or periodic discharges (Figure 2).

MRI and DWI sequence (axial cuts) revealing gyriform pattern of diffusion restriction in (a) basal ganglia (caudate and putamen) and bilateral thalami, (b) caudate and along the cortex, and (c) bilateral medical cerebral hemispheres.
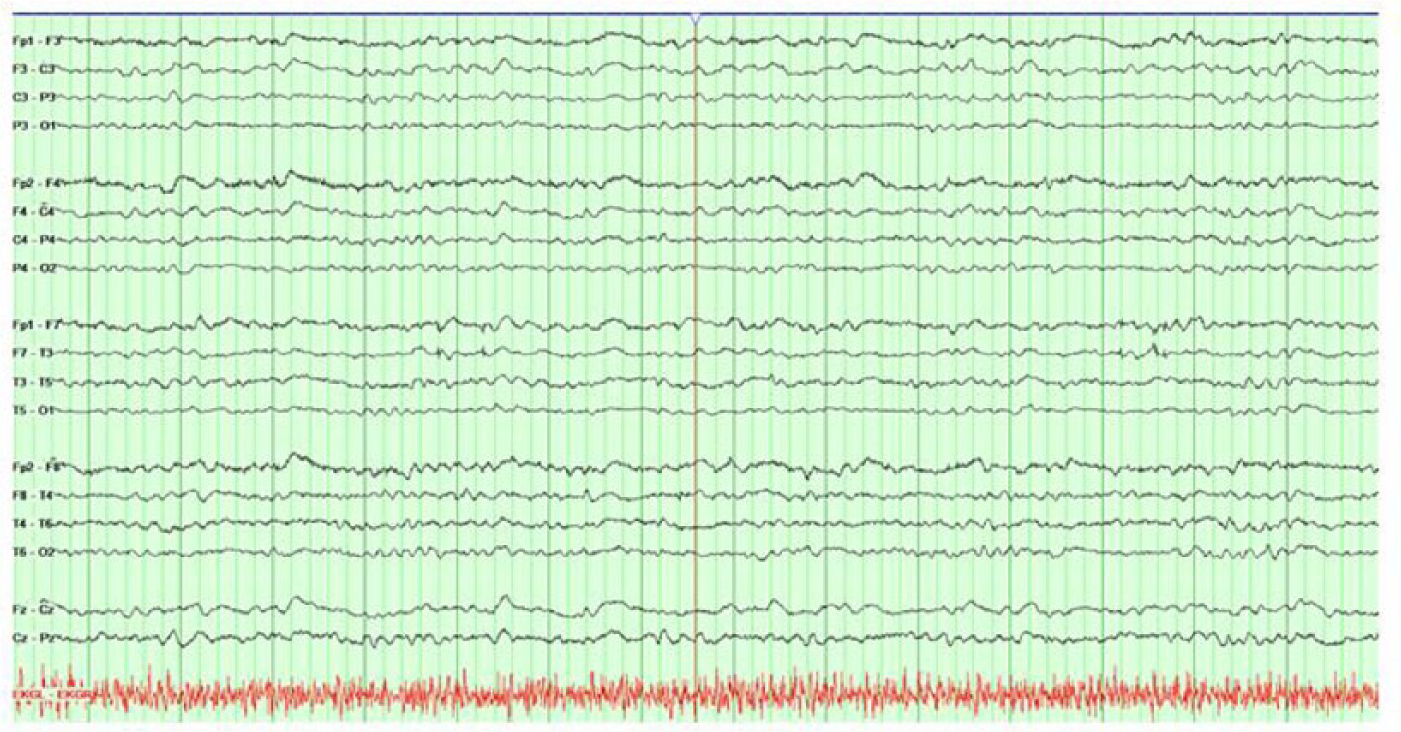
Continuous EEG monitoring, with noted background activity consisting predominantly of 4–6 Hz activity, with initial waxing and waning periods of relative suppression for 1–3 s (not shown here) when the recording was initiated. There was no clear evidence for an occipital dominant rhythm, and definite delineation between sleep and wakefulness was not possible. There was no focal slowing, no interictal discharges, and no electrographic seizures identified.
Discussion
Changes in memory, concentration, attention, and motor skills are not uncommon in patients with HIV and have long been recognized as nervous system sequelae of the virus itself, but these symptoms can also be caused by a myriad of other conditions. 12 Given the broad differential diagnosis and associated therapeutic implications, reaching an accurate diagnosis is critical. With the patient’s long-standing (>two decades) HIV infection, one possible explanation for his symptoms is HIV-associated neurocognitive decline, which is classically characterized by subcortical dysfunction, attention and concentration impairment, mood disorder, and impaired psychomotor precision.12–14 However, his HIV was well controlled, with a CD4 count >400/µL, and an undetectable viral load for most of the last 15 years made this less likely. Another potential etiology of his neurocognitive symptoms is a subacute to chronic CNS infection. As part of his work-up, we performed a lumbar puncture (Table 1), which should be performed in any immunocompromised patient with an alteration of consciousness. However, these CSF studies were unremarkable. While this patient did have aspiration pneumonia, this infection was felt to follow rather than precede the patient’s change in mental status. A third potential explanation for his subacute neurocognitive decline is structural abnormalities of the brain, such as stroke or tumor. Brain imaging revealed no clear structural lesion, but did show a peculiar pattern of diffusion restriction in the deep brain nuclei (basal ganglia, putamen, and globus pallidus) and a pattern of ribbon-like gyriform diffusion restriction along the cortices (Figure 1). Such findings are non-specific and can be seen in variety of disease processes, including anoxic brain injury, carbon-monoxide poisoning, heavy metal poisoning, stroke, vasculitis, reversible posterior leukoencephalopathy syndrome, inherited metabolism/neurodegenerative disorders, and prion disease.15,16 All of these conditions except prion diseases were excluded based on laboratory work-up and clinical phenotype.
Summary of work-up during patient’s hospitalization.

WBC: white blood cells; JC: John Cunningham; TSH: thyroid-stimulating hormone; BUN: blood urea nitrogen; AST: aspartate transaminase; ALT: alanine transaminase; EBV PCR: Epstein–Barr virus polymerase chain reaction; CSF: cerebrospinal fluid; VDRL: venereal disease research laboratory; CMV: cytomegalovirus; HSV: herpes simplex virus; RT-QuIC: real-time quaking-induced conversion.
Numerous criteria have been suggested for the diagnosis of CJD,17–19 and although autopsy is the gold standard for diagnosis, characteristic clinical and laboratory features are often sufficient for a diagnosis of “probable” or sporadic CJD (sCJD). Supportive diagnostic modalities such as MRI abnormalities may vary with the clinical syndrome and the molecular subtype. For example, patients with sCJD who demonstrate increased T2 signal in the caudate and putamen are likely to develop early dementia and increased mortality.2,20 Diffusion weighted imaging (DWI) changes are overall more sensitive than T2-flair for the detection of CJD-related lesions on brain MRI, particularly for cortical findings. Our patient exhibited classical radiologic findings seen in sCJD. Electroencephalography findings may provide additional supportive evidence for CJD.21–24 Characteristic patterns include periodic synchronous biphasic or triphasic sharp wave complexes, diffuse slowing, or periodic lateralized discharges in some cases.21–23 Our patient’s EEG lacked the classical periodic discharges, which are neither sensitive nor specific to CJD, but did exhibit a pattern of diffuse slowing (Figure 2). Laboratory markers may serve as adjuvant markers of prion disease.18–20,25 The use of 14-3-3 protein, neuronal specific enolase (NSE), and S100 has all been described. These proteins are released into CSF with neuronal cell damage; however, none of these tests have absolute sensitivity or absolute specificity for prion disease.26–31 Recently, an ultrasensitive test in CSF by real-time quaking-induced conversion (RT-QuIC®) has proved very useful. Several groups have achieved sensitivity >80% and specificity of 100% with the use of this lab assay.32–34
In patients presenting with rapid cognitive decline with associated neuroimaging abnormalities suggestive of prion disease, it is important to distinguish CJD and other prion-related neurodegenerative processes from dementia. Alzheimer’s dementia and frontotemporal dementia may be associated with a rapid atypical course that mirrors prion disease.35,36 Ataxia and Parkinsonism can also present with progressive supranuclear palsy and multiple system atrophy.35,36 However, even with the most rapidly progressive dementias, virtually none result in the death of the patient within weeks to months of symptom onset. The latter is a more typical of CJD. However, in situations where a patient may present with confounding picture that is incongruent for definite clinical diagnosis of CJD, the utility of confirmatory testing with high specificity, such as RT-QuIC, may be most helpful. Based on the patient’s presenting signs and symptoms, brain imaging findings, and lack of another obvious explanation for the patient’s rapid neurocognitive decline, the possibility of prion disease was entertained; CSF markers confirmed our suspicion for CJD. Eventually, and after discussion with the patient’s wife, supportive measures were instituted and the patient was transferred to hospice care.
Interestingly, this patient’s HIV status, despite well controlled, may have, at least theoretically, also predisposed to prion disease. Despite prions and HIV being very different pathogens, their requirement for cholesterol for propagation and infection may be a common element. 37 Regulation of adenosine triphosphate (ATP)-binding cassette transporter type 1 (ABCA1) functionality by its mislocalization is a well-established phenomenon exploited by pathogens, such as HIV, and the effect of prions on ABCA1 and lipid rafts is very similar to what has been found with HIV.37,38 The pathway by which prion infection post-translationally modifies cholesterol efflux by displacing ABCA1 from lipid rafts is not dissimilar to that used by HIV, which also requires cholesterol for its replication. 38 This suggests that while prions and HIV viruses are very different, they may involve the same cellular mechanism of cholesterol metabolism.
Conclusion
This case highlights spontaneous CJD as a rare terminal illness in the setting of chronic HIV. While the patient’s long-standing HIV initially suggested his symptoms could be due to HIV-associated neurocognitive decline, we determined with a high degree of certainty that he had spontaneous CJD based on clinical, radiographic, laboratory, and EEG findings. This was subsequently confirmed on autopsy. To the best of our knowledge, this is the first report of a patient with chronic and previously well-controlled HIV infection succumbing to a prion disease. Despite the very different epidemiology and pathophysiology of HIV and sCJD, this case does raise questions on whether certain host genetic factors could predispose to both conditions. 39 In the interim, no known causal link between HIV and spontaneous CJD has yet been firmly established.
Footnotes
Acknowledgements
The authors were responsible for the medical care and management of the patients described in this manuscript. All authors agree to the conditions outlined in the Authorship and Contributorship section of the information for authors. All authors contributed to the medical care of this patient. All contributed to the manuscript preparation and revision. All authors read and approved the final manuscript. No statistical analysis was performed.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article. Consent was obtained from next of kin to publish this report.