
Abstract
We report a 13-year-old youth who initially presented with the typical rash of Henoch–Schonlein purpura followed a month later by a nephrotic syndrome and hematuria. Renal biopsy revealed crescentic IgA nephropathy. The patient was aggressively treated with steroids leading to a remission of his nephrotic syndrome. Three years after his initial presentation, he developed bloody diarrhea and Crohn’s disease was diagnosed.
Introduction
Henoch–Schonlein purpura (HSP) is a syndrome characterized by the tetrad of cutaneous purpuric rash, arthritis, nephritis and gastrointestinal manifestations.1,2 HSP nephritis forms part of the spectrum of IgA nephropathy (IGAN). Secondary IGAN has been linked to liver disease, in particular alcoholic cirrhosis, and to mucosal inflammation. An increasing literature has reported an association between IGAN and inflammatory bowel disease (IBD). In fact, in a case series of kidney biopsies in IBD patients, IGAN proved to be the most common diagnosis. 3 Here, we report a patient in whom HSP nephritis was antecedent to the diagnosis of his IBD (Crohn’s) by 3 years. Although gastrointestinal symptomatology may be seen in HSP patients, when prolonged the possibility of co-existent IBD should be borne in mind. In our patient, the aggressive treatment administered for his renal injury may have masked or delayed the onset of IBD.
Case report
Publication of this case report does not require ethics approval in our institution. The patient’s parents have given written informed consent for it to be published in a scientific journal.
A previously healthy 13-year-old Caucasian youth presented with a petechial rash over the lower limbs and buttocks. The rash had first appeared 3 days prior to admission in the beginning of 2013. There was no other significant symptomatology. Patient’s weight was 74 kg, height 1.65 m, body mass index 27.18 kg/m2 and blood pressure 115/66 mmHg. Apart from the palpable petechial rash, the remainder of the physical examination was non-contributory. Laboratory data showed a hemoglobin of 12.1 g/dL with a mean corpuscular volume (MCV) of 75.9 fL, a mean corpuscular hemoglobin (MCH) of 25.7 pg, white blood cell (WBC) count of 6300 L−1 and platelet count of 218/µL. Serum creatinine was 0.7 mg/dL. A serological panel inclusive of anti-nuclear antibodies (ANA), anti-neutrophil cytoplasmic antibodies (ANCA) and complement levels (C3, C4) was negative. On dipstick urinalysis, there was trace blood and protein. Although no skin biopsy was performed, HSP was diagnosed based on the typical rash and its distribution. The patient was discharged for follow-up outpatient care.
A month later, he was re-admitted with a full blown nephrotic syndrome (proteinuria of 11.3 g/24 h, serum albumin 2.8 g/dL and cholesterol 244 mg/dL). Urine sediment showed 10–15 red blood cells per high power field (RBC/HPF). Serum creatinine remained at 0.7 mg/dL. A renal biopsy was performed (findings detailed below).
Three years later, he presented with bloody diarrhea for which colonoscopy was performed. There was no associated exacerbation of his renal disease.
Hospital course
On light microscopy, although there were only four glomeruli, they all showed the same appearance. This consisted of focal mesangial expansion with mesangial cell proliferation and a thickened glomerular basement membrane (GBM). In two of the glomeruli, a fibrocellular crescent was seen (Figure 1). Immunofluorescence was strongly positive for IgA along the GBM and in the mesangium (Figure 2). On electron microscopy, scattered mesangial electron dense deposits were seen.

Glomerulus showing focal mesangial proliferation and a fibrocellular crescent in the lower part of the glomerulus (H&E stain).
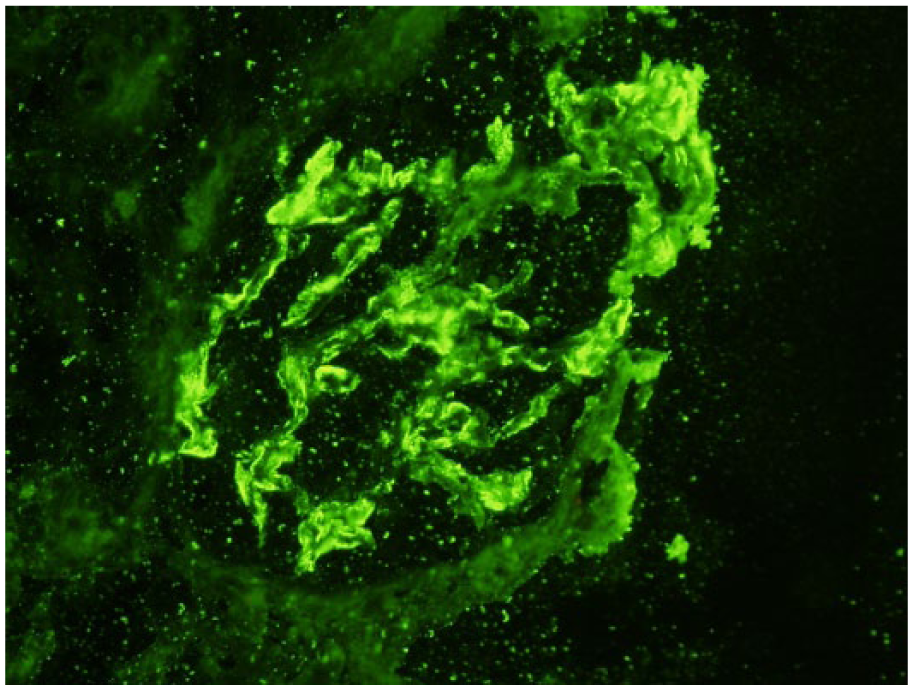
Immunofluorescence with IgA showing strong positive staining along the GBM.
Treatment was initiated with pulse steroids (Solumedrol) 1 g/day for 3 days followed by oral prednisone at 60 mg/day and fish oil at 3 g/day. Steroids were tapered over a period of 1 year at the end of which proteinuria had decreased to 431 mg/24 h and over the ensuing year to <150 mg/24 h with a return of serum albumin to 4.0 g/dL.
Three years after the renal biopsy, colonoscopy revealed widespread inflammatory lesions extending from rectum to cecum and involving the terminal ileum. Ulcerative lesions were clearly delineated and surrounded by normal mucosa forming skip lesions. The intestinal mucosa showed a nodular surface giving a cobblestone appearance. Biopsies of the colon and terminal ileum showed a similar appearance, namely, a heavy cellular infiltrate consisting of both lymphocytes, plasma cells mixed with neutrophils and eosinophils. There were multiple crypt abscesses and foci of cryptitis (Figure 3). No granulomata were seen nor was there any evidence of vasculitis. Crohn’s disease was diagnosed and the patient treated with 5-aminosalicylic acid with good response.
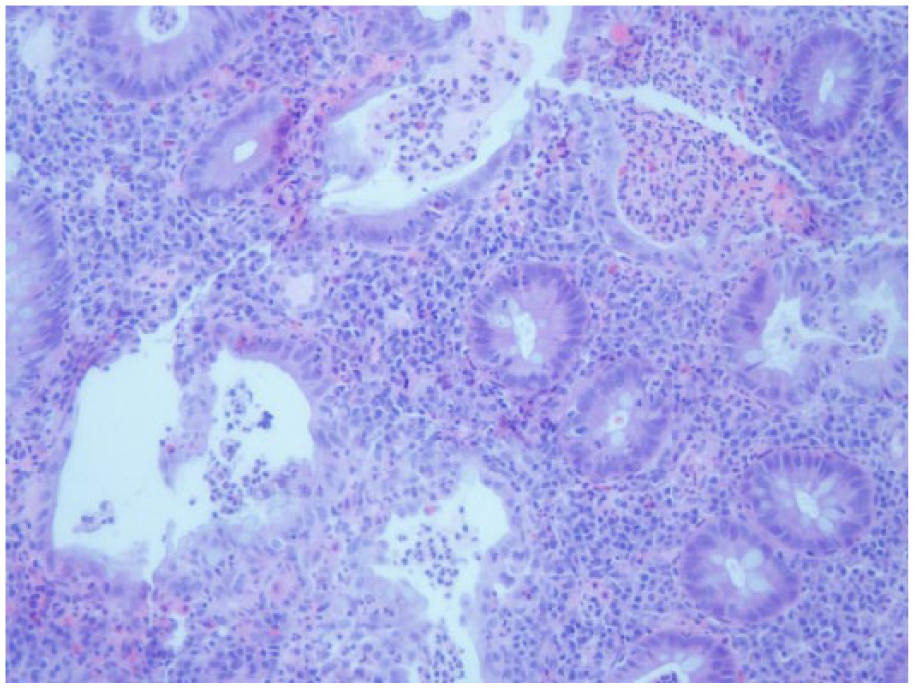
Colon biopsy showing heavy cellular infiltrate with crypt abscesses.
Discussion
HSP is a syndrome characterized by the tetrad of cutaneous purpuric rash, arthritis, nephritis and gastrointestinal manifestations (pain and bleeding). It is a small vessel vasculitis mediated by IgA immune complex deposition in vessel walls.1,2 Although it may occur in adults, it is primarily a disease of young children and is actually the most common systemic vasculitis encountered in the pediatric population. HSP nephritis falls into the spectrum of IGAN and clinically may range from asymptomatic microscopic hematuria and/or mild proteinuria to a nephrotic syndrome and rapidly progressive crescentic renal failure. Our patient demonstrated the typical purpuric rash of HSP and a month later developed a full blown nephrotic syndrome. This course of events is in keeping with the literature which reports that the rash usually precedes renal involvement. 4 In view of the severe nephrosis and the renal biopsy histological findings (despite an inadequate number of glomeruli), crescentic IGAN was diagnosed and aggressively treated. The combined steroid/fish oil treatment resulted in a full remission. Notably, between 35% and 60% of cases of HSP manifest gastrointestinal symptomatology. These include colicky abdominal pain, nausea, vomiting and bleeding. 5 In our patient, no gastrointestinal symptoms were present at the time HSP nephritis was diagnosed.
Rectal bleeding developed 3 years after HSP nephritis was diagnosed. Colonoscopy and biopsy demonstrated the typical appearance of Crohn’s disease. Urological complications of IBD may be seen in up to 25% of patients and include calcium or urate stones, urinary tract infections and enterovesical fistulas. 5 Renal parenchymal disease is, however, a rare occurrence. Nevertheless, an increasing number of reports have documented an association between IBD and IGAN. In a recent retrospective case series of kidney biopsies in patients with IBD (45 cases of Crohn’s and 38 cases of ulcerative colitis), 20 of 83 (24%) biopsy specimens showed IGAN followed by 16 (19%) with interstitial nephritis. 3 IGAN was, therefore, the most common diagnosis and its prevalence was significantly higher compared with all non-IBD kidney biopsies. Four of the 20 cases had a concurrent vasculitic rash confirmed by skin biopsy in two of the cases as leukocytoclastic vasculitis. Since the first description of IBD-associated IGAN by Hubert et al. 6 in 1984, 19 subsequent case reports have documented such an association. In the majority of these patients, IGAN developed during onset or exacerbation of IBD and at least partial clinical remission of the kidney disease was obtained upon successful treatment of bowel inflammation. In our patient, no renal relapse was evident at the time Crohn’s disease was diagnosed.
The increased prevalence of IGAN in IBD may imply a common pathogenesis. Given the important immunologic role of IgA in the defense against environmental and microbiologic antigen exposure at mucosal sites, IBD-associated IGAN is within the realms of possibility. The occurrence of IGAN in IBD is therefore likely to represent a complex interplay involving mucosal inflammation, increased mucosal permeability and thus loss of antigenic exclusion, chronic immune stimulation and dysregulated IgA production and transport. Both intestinal inflammation and IGAN were shown to be T-cell mediated. 7 Genetic susceptibility is also implicated as an association with HLA-DR1 has been reported in both IGAN and IBD. 8 The above data are in favor of a common pathogenic link operative in both diseases.
Conclusion
This case describes the association of HSP inclusive of nephritis with IBD, specifically Crohn’s disease. Gastrointestinal symptomatology is seen in a not-infrequent percentage of HSP patients. 4 When, however, these symptoms are prolonged, the possibility of co-existent IBD should be borne in mind. In our patient, the aggressive treatment administered for his renal condition may have actually masked or delayed the onset of IBD. The commonest glomerular pathology in IBD is IGAN and its prevalence in IBD exceeds that found in non-IBD patients.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.