
Abstract
Congenital lymphedema is a rare type of primary lymphedema occurring at birth or developing shortly later. Primary lymphedema can be classified according to whether it is familial or sporadic. The primary congenital familial lymphedema is also known as Milroy’s disease. Majority of primary cases are sporadic type. Chronic lymphedema can be secondary to infections, surgery with lymph node excision, trauma, lymphadenectomy, radiotherapy, filarial infection, and so on. It is recognized that a variety of malignant tumors can arise in chronic congenital or acquired lymphedema; the most documented associations are lymphangiosarcoma, basal cell carcinoma, lymphoma, malignant melanoma, and Kaposi’s sarcoma. A total of 13 cases of squamous cell carcinoma arising from chronic (primary or acquired) lymphedema have been reported, and only 3 cases of congenital lymphedema presented with squamous cell carcinoma as reported. A 32-year-old young male presented with chronic unilateral left lower limb lymphedema of 28 years duration. In addition, he had a 3-month history of a fungating cutaneous lesion on the lateral side measuring 2 cm × 1 cm in size. Fine-needle aspiration cytology was performed on the later mass, and a diagnosis of angiosarcoma was made. At histopathology, the appearances did not confirm angiosarcoma. However, an impression of carcinoma was made as squamous cells were observed in sheets. Immunohistochemistry was performed using markers for CD31, factor VIII (FVIII), and MiB. The epithelial marker cytokeratin was positive for squamoid cells and MiB index of 75%. The vascular markers FVIII and CD31 were negative, thus ruling out angiosarcoma. The final diagnosis was given as infiltrating squamous cell carcinoma in chronic lymphedema.
Introduction
Squamous cell carcinoma (SCC) arising from chronic lymphedema is rare, and only 13 cases have been reported in the literature so far. The most common form of primary lymphedema is the sporadic type. 1 Familial forms represent only 5%–10% of primary lymphedema. Primary lymphedema appearing at birth is known as Milroy’s disease, and when it appears at puberty or post puberty, it is known as Meig’s disease. 2 Secondary lymphedema is encountered more often and is seen associated with conditions such as infections, surgery with lymph node excision, radiotherapy, and so on. It affects 15%–20% of women receiving axillary dissection and/or radiotherapy as part of their breast cancer treatment. 3 Lymphedema may be complicated by the development of many cutaneous malignancies, and majority of them are angiosarcoma, 4 Kaposi’s sarcoma, 5 lymphoma, 6 basal cell carcinoma, 7 and so on. Of the reported cases in the literature, three cases have shown their origin from congenital chronic lymphedema.8–10 This is the fourth case of SCC seen on a 32-year-old male with chronic congenital lymphedema (CL) presenting at the age of 4 years. The objective of this study is to define the evolution, pathogenesis, and presentation of SCC arising from long-standing congenital sporadic chronic lymphedema.
Case report
A 32-year-old man was referred from another hospital with massive swelling of the left lower limb since 28 years. The swelling was present since childhood and gradually increased over time. Only 6 months back, he noticed multiple bullae arising from the lymphadematous limb measuring 0.5 cm × 0.5 cm to 3 cm × 3 cm on the lateral upper aspect of the leg; few of them had ruptured causing painful superficial ulcers over 2–3 months exuding serosanguinous fluid. One ulcer showed a vegetative growth (Figure 1). On examination, the skin was indurated in several places with red-to-brownish macules and papules showing evidence of healing ulceration at one focus. Inguinal lymph nodes were not palpable. There was no familial history of lymphedema. The patient was wrongly diagnosed for filarial lymphedema 10 years earlier, for which treatment was given. There was no response to the adequate antifilarial treatment. Repeated buffy coats did not show any filarial organism, which made us suspicious for the other cause of lymphedema. He also narrated that he frequently washed the limb with antiseptic solution for the past 20 years. He was referred to Department of Pathology for fine-needle aspiration cytology (FNAC). The ulcers ranged in size from 1 cm × 2 cm to 3 cm × 2 cm. The procedure was performed from the thickened skin around the ulcers, and the slides were fixed and stained with Papanicolaou (PAP) and May–Grünwald-Giemsa (MGG) stains.

Ulceration of lymphadematous limb.
Smears showed moderately cellular smear that showed a vasoformative pattern with bunches of radiating plump and pleomorphic plump epithelial cells in clusters, syncytial pattern and singly scattered, and occasionally showing vacuolated cytoplasm. The cells were large with dark hyperchromatic nuclei and moderate eosinophilic cytoplasm. The diagnosis of a vasoproliferative lesion, probably angiosarcomas, was considered on FNAC (Figure 2). Biopsy was also taken from the same site and sent to histopathology for processing. The sections showed skin tissue with focal ulceration of the epidermis, and the upper dermis showed large vascular spaces lined by flattened to mildly plump endothelial cells. The lower dermis showed collection of prominent neoplastic squamoid cells arranged in small groups and islands with wide areas of necrosis infiltrating into the subcutis. The squamous cells were plump and polygonal and had vesicular nucleus and moderate to abundant cytoplasm (Figure 3, 4 and 5). A focal nest of tumor cells showed a squamous pearl, giving a provisional diagnosis of poorly differentiated SCC/angiosarcoma, with further confirmation by immunohistochemistry. The vascular markers factor VIII (FVIII) and CD31 were found to be negative (Figure 6). The tumor cells were positive for cytokeratin and MiB (Figures 7 and 8). A diagnosis of infiltrating SCC arising from chronic sporadic CL was confirmed.
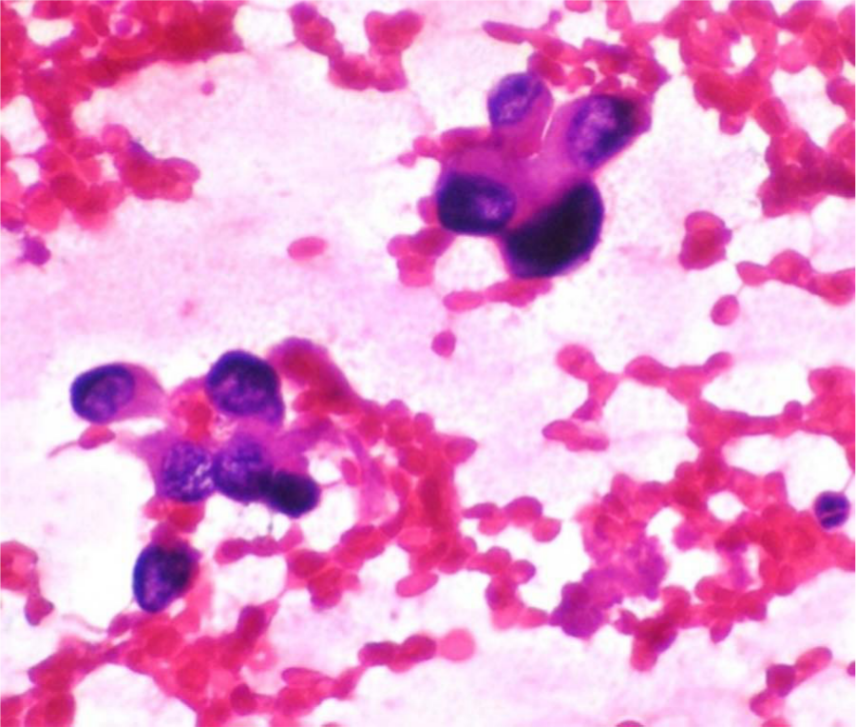
Radiating plump epithelioid cells.

Dermis showing spaces mimicking a squamoid vascular lesion.
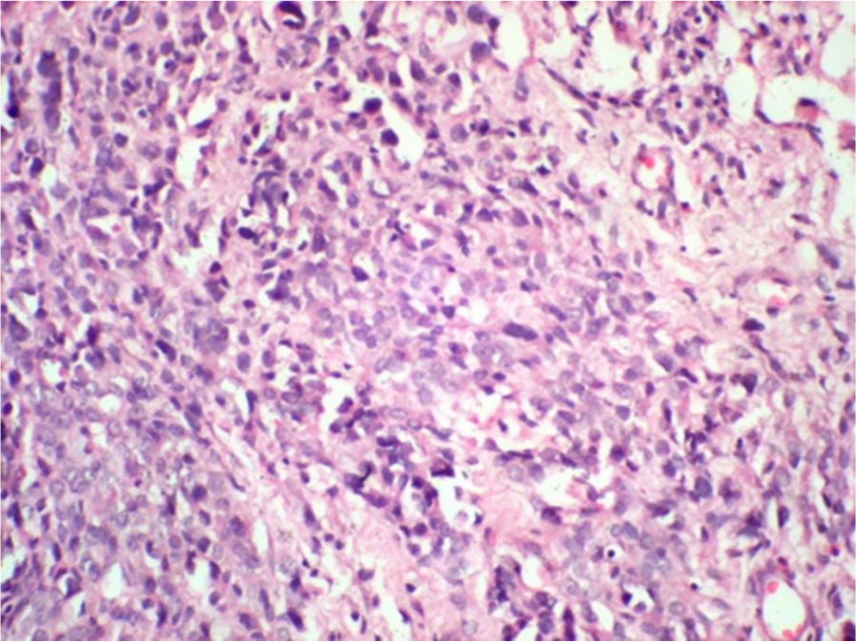
Pleomorphic squamoid cells in islands.

Lower dermis showing pleomorphic cells with a keratin pearl.

CD31 negative.

Cytokeratin positivity.

75% positivity of MiB.
Discussion
Primary lymphedema may arise from an intrinsic abnormality, for example, genetically determined aplasia, hypoplasia, or dysfunction of lymphatic vessels. 11 The long-standing lymphedema is characterized by trapping the fluid in the skin and subcutaneous tissue, plasma proteins, and other macromolecules. 11 In the present case, it is a classical example of sporadic CL. The patient presented with the swelling of the right leg at the age of 4 years. There was no other acquired reason like infection, trauma, surgery, or venous disease for the development of the same. He did not belong to nor had he travelled to any coastal region of our country where filariasis is prevalent. In most cases, lymphedema primarily affects the lower limbs, starting with the feet, but it can also affect the upper limbs. Females are twice as likely as males to have CL, but in this case, it was a male patient. 12
The chance of occurrence of SCC arising from long-standing primary CL is rare. Only three cases have been described in the literature, and there has been little emphasis on predisposing dermatological conditions and factors responsible for SCC in chronic lymphedema.8–10
Many theories have been proposed for carcinogenetic predisposition of chronic lymphedema. The reason is the obliteration of lymphatic channels in the adjacent tissue by scarring of ulcer, hence antigens specific to the tumor cannot reach the lymph nodes. Therefore, the normal immunosurveillance mechanisms related to these regional nodes cannot react with the antigens. This allows primary tumor growth unrestricted by the usual immunologic mechanisms. Chronic lymphedema is believed to be an independent and important factor in tumor development, and inherited susceptibility to carcinogenic stimuli is also thought to play a role. In our case, the heightened susceptibility to local irritation by continuous exposure to the antiseptics over a period of 20 years might have been carcinogenic and played an important role in the formation of SCC. The presence of chronic ulceration may act as a promoter factor in the carcinogenesis of SCC. The importance of a random genetic mutation occurring in hyperplastic tissue can also be hypothesized. Factors implicated in the pathogenesis observed in the literature are verrucous hyperplasia, ultraviolet therapy for psoriasis, dystrophic epidermolysis bullosa, vitiligo, and epidermodysplasia verruciformis.10,13 None of these conditions existed in the present case.
Complications of chronic limb lymphedema include recurrent cellulitis, while tumoral complications are very rare and the most described is Stewart–Treves lymphangiosarcoma. Kaposi Sarcoma, basal cell carcinoma, B-cell lymphoma, and malignant melanoma are very unusual and reported.4–7 Stewart–Treves syndrome refers to the development of lymphangiosarcoma in a patient with chronic post mastectomy lymphedema. 14 In a comprehensive study done by Armer et al., 15 the authors report the incidence of symptomatic lower extremity lymphedema to be as high as 36% in women with a history of gynecologic cancer, with the highest rates occurring in women treated for vulvar cancer.
The cytological features of angiosarcoma are variable. Boucher et al. 16 stated that cytologic features of angiosarcomas revealed by fine-needle aspiration (FNA) are heterogeneous and may overlap with those of other sarcomas and poorly differentiated carcinomas. In the present case, FNA specimen exhibited highly atypical loosely cohesive epithelioid cells with high nuclear/cytoplasmic ratios, vesicular irregular chromatin, and conspicuous nucleoli and moderate amount of cytoplasm. Tumor cells were seen apparently lining vascular channels. This finding coupled with history of long-standing lymphedema and swelling of the leg prompted the diagnosis of angiosarcoma. This was, however, disproved by markers.
The histopathology showed features of poorly differentiated SCC. Nappi et al. 17 illustrated in their report six cutaneous SCCs that closely simulated angiosarcomas on conventional histologic examination. There have been case reports in which squamous cells differentiating into acantholytic areas may mimic types of adenocarcinoma or sweat gland carcinoma or may form a pseudo vascular pattern resembling angiosarcoma. In contrast to the voilaceous and papular appearance of angiosarcoma, SCC presents itself as a discrete cutaneous ulcer. In the present case, SCCs presented with ulceration on voilaceous papules and macules.
In patients with lymphedema, physical/medical treatment plays a pivotal therapeutic role. The medical treatment for SCC includes complete surgical resection of the tumor with free margins, along with radiation therapy and chemotherapy used as adjunctive approaches. 18 External compression by multilayer lymphedema bandaging has also proved to be useful to combat lymphedema.
Conclusion
SCC arising in CL is very rare and should be considered in the differential diagnosis of malignancy arising as ulcerative growth from lymphadematous skin. Localized long-term application of antiseptics may induce cutaneous malignant lesions. The cytology of SCC can mimic that of angiosarcoma. We suggest that SCC may be distinguished from angiosarcoma of the skin by attention to its clinical features and appropriate immunohistochemical slides.
Footnotes
Declaration of conflicting interests
The authors declare no conflict of interest in preparing this article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.