
Abstract
Objective:
We aimed to investigate the functional alterations caused by pathogenic variants in the FOXL2 gene, a forkhead transcriptional factor.
Methods:
This study is an experimental research with a duration from January to September 2022. We selected six variants for analysis, including a double missense variant, c.150C>G (p. Asp50Glu) and c.326A>T (p. Asn109Ile); three deletions, c.411_412del (p. Met137Ilefs101), c.533_542del (p. Val178Alafs90), and c.684delA (p. Ala229Leufs43); a nonsense variant, c.214G>T (p. Glu72); and a duplication, c.663_692dup (p. Ala225_Ala234dup). We constructed expression vectors containing these variants and transfected them into HeLa cells. Confocal microscopy was used to observe the subcellular localization of the expressed proteins. We evaluated gene expression using dual luciferase reporter assays and quantitative PCR.
Results:
Proteins expressed by vectors with deletion variants were predominantly localized to the nucleus, while those with the double missense variant exhibited diffuse expression throughout the cell. Proteins from nonsense and duplication variants localized to the cytoplasm. Luciferase activity assays revealed that proteins encoded by the p. Ala229Leufs43, p. Glu72, and p. Ala225_Ala234dup variants significantly diminished the inhibitory effects on the transcription of the StAR gene. Additionally, all proteins encoded by indel and nonsense variants, except for the double missense variant, demonstrated a marked reduction in their inhibitory effects on CCDN2 and INHBB gene expression.
Conclusions:
The double missense variant does not exert a superimposed inhibitory effect on gene expression. Despite differences in subcellular localization, all mutant proteins produced by these variants likely interfere with downstream gene expression through a shared pathway. Furthermore, mutant FOXL2 proteins may disrupt ovarian development via multiple pathways, extending beyond their impact on StAR gene expression.
Introduction
The FOXL2 (forkhead transcriptional factor 2; OMIM 605597) gene is a single-exon gene that encodes a 376-amino-acid protein belonging to the Forkhead Box (Fox) transcription factor superfamily. Structurally, FOXL2 contains a DNA-binding forkhead domain (FHD) at the N-terminus, spanning approximately 100 amino acids from codons 54 to 148. At the C-terminus, it features a polyalanine tract consisting of 14 amino acids, from codons 221 to 234. 1
The FOXL2 protein is predominantly expressed in developing eyelids, somatic cells of the embryonic ovary, and granulosa cells within ovarian follicles. It serves as a critical transcriptional regulator, playing a key role in embryonic eyelid and ovarian development, as well as in the growth and function of female gonads. Additionally, FOXL2 participates in regulating steroid metabolism, detoxifying reactive oxygen species, and modulating inflammation in the female reproductive system.2–4
FOXL2 exerts its effects on sexual differentiation and follicular development by regulating a network of downstream target genes. These include SRY (sex-determining region Y), INHBB (inhibin subunit beta B), SOX9 (SRY-box transcription factor 9), CYP11A1 (cytochrome P450 family 11 subfamily A member 1), CCND2 (cyclin D2), PTGS2 (prostaglandin-endoperoxide synthase 2), BCL2A1 (BCL2-related protein A1), IER3 (immediate early response 3), IFNB1 (interferon beta 1), IL12A (interleukin 12A), and STAR (steroidogenic acute regulatory protein). These genes regulate various processes, including sex determination, granulosa cell differentiation and proliferation, and steroid hormone biosynthesis.5–8
Mutations in the FOXL2 gene can lead to premature ovarian failure (POF) and an autosomal dominant disorder known as blepharophimosis–ptosis–epicanthus inversus syndrome (BPES; OMIM 110100). BPES is characterized by narrow palpebral fissures (blepharophimosis), drooping eyelids (ptosis), and an inward and upward fold of skin from the lower eyelid (epicanthus inversus). 4 To date, researchers have identified over 100 FOXL2 mutations associated with POF and BPES, though the precise molecular mechanisms underlying these conditions remain unclear.
BPES is classified into two subtypes: Type I, which includes POF, and Type II, which does not. Each subtype appears to be associated with specific genotypes. For example, missense mutations may result in either Type I or Type II. Deletions within the polyalanine tract are linked to Type I, while expansions of the polyalanine tract are associated with Type II. 3
In our previous studies, 9 we identified six specific FOXL2 variants: a double missense mutation, c. 150C>G (p. Asp50Glu) and c.326A>T (p. Asn109Ile); a nonsense mutation, c.214G>T (p. Glu72*); three deletions, c.411_412del (p. Met137Ilefs101), c.533_542del (p. Val178Alafs90), and c.684delA (p. Ala229Leufs43); and a duplication, c.663_692dup (p. Ala225_Ala234dup). Patients with each variant of FOXL2 show blepharophimosis-ptosis-epicanthus inversus syndrome, which is characterized by narrow palpebral fissures, bilateral ptosis, and epicanthus inversus. However, we were not able to establish genotype-phenotype correlations for these patients due to the lack of adult affected females in these families (Supplemental Table 1). Among these variants, c.214G > T (p. Glu72), c.411_412del (p. Met137Ilefs101), and c.533_542del (p. Val178Alafs90) occurred within the prepolyalanine region, either with or without an intact FHD. These mutations are predicted to produce truncated proteins lacking the polyalanine tract. The c.684delA (p. Ala229Leufs*43) variant is predicted to produce a truncated protein that retains part of the polyalanine tract. In contrast, the c.663_692dup (p. Ala225_Ala234dup) variant is expected to result in an expanded polyalanine tract (Figure 1). In this study, we examined the functional significance of the FHD and the polyalanine tract in FOXL2. Additionally, we investigated whether the double missense variants c.150C > G (p. Asp50Glu) and c.326A > T (p. Asn109Ile) exhibit a superimposed effect on functional alterations compared to single missense variants.

Schematic of pathogenic variants. The orange box refers to the forkhead domain, the purple box refers to the Poly-Ala tract, and the green box refers to changes in proteins.
Methods
This study is an experimental research with a duration from January to September 2022.
Construction of expression vectors
We amplified the full-length FOXL2 open reading frame using PCR and cloned it into pEGFP and pcDNA3.1-EF1α vectors, generating the EGFP-FOXL2-WT and pcDNA3.1-EF1α-FOXL2-WT plasmids, respectively. Mutant vectors were constructed using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) with the wild-type plasmid as the template. The primers used for mutagenesis are listed in Supplemental Table 2. The mutagenesis reaction was performed in a 50 µL volume containing template DNA and other required reagents, following the kit’s protocol. The cycling conditions included an initial denaturation step at 94°C for 2 min, followed by 16 cycles of denaturation at 96°C for 15 s, annealing at 65°C for 15 s, and extension at 68°C for 4 min, with a final extension at 68°C for 1 min.
The product was digested with diphosphopyridine nucleotide (Dpn I) and incubated at 37°C for 1.5 h. We used 5 µL of the digested product to transform E. coli. To create the reporter plasmid pcDNA3.1-StAR-Luciferase-SV40-Puro, we replaced the CMV promoter with the StAR promoter. The primers for the StAR promoter were as follows:
StAR-F: TTCGAACCTAGTTAGATGTTTCACCATGTTGGCCA
StAR-R: ACTGATGCTAGCTGTTTCCTGGCAAATGTGGCAGTGGT
All expression constructs were sequenced to confirm the presence of the target fragments and to exclude any additional PCR-induced variations.
Cell culture and transfection
Hela cells (ATCC CCL-2) were cultured in petri dishes containing DMEM (Gibco, CA, USA) supplemented with 10% fetal calf serum (Gibco–Invitrogen, Grand Island, NY, USA) and 1% penicillin/streptomycin. The cells were evenly seeded into a 6-well plate with 2 mL per well and incubated at 37°C. Once the cells reached 70%–80% confluence, each well was transfected with 2 µg of EGFP-FOXL2-WT, EGFP-FOXL2-MT, or EGFP-FOXL2-NC using 3 µL of Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). After 48 h, cells were fixed with 4% paraformaldehyde for 30 min, and the nuclei were stained with 4ʹ,6-diamidino-2-phenylindole (DAPI) at a 1:5000 dilution. All transfection experiments were performed in triplicate. Localization of the wild-type and mutant FOXL2 fusion proteins was visualized using a confocal laser scanning microscope (A1R, Nikon, Tokyo, Japan).
Dual-luciferase report assays
For luciferase assays, Hela cells in the 6-well plates were co-transfected with pcDNA3.1-StAR-luciferase-SV40-Puro and pcDNA3.1-EF1α-FOXL2-WT, pcDNA3.1-EF1α-FOXL2-MT, or pcDNA3.1-EF1α-FOXL2-NC using 3 µL of Lipofectamine 2000 reagent. Each well received 1 µg of the target vector and 0.5 µg of the pRLuc-TK vector as an internal control to normalize transfection efficiency. An empty vector served as the negative control. After 48 h, cells were lysed, and the supernatant was collected following centrifugation. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) on a Lumimark luminometer (Bio-Rad Laboratories, Hercules, CA, USA) and expressed as relative light units (RLUs). All experiments were conducted in triplicate.
Quantitative real-time PCR (qPCR)
We selected eight genes—INHBB, CYP11A1, CCND2, PTGS2, BCL2A1, IER3, IFNB1, and STAR—to evaluate the impact of FOXL2 on their expression. Total RNA was extracted from cells transfected with pcDNA3.1-EF1α-FOXL2-WT, pcDNA3.1-EF1α-FOXL2-MT, or pcDNA3.1-EF1α-FOXL2-NC using TRIzol reagent (Invitrogen, Carlsbad, USA), according to the manufacturer’s instructions. cDNA synthesis was carried out using the Superscript IVIV Kit (Invitrogen, Carlsbad, USA), and qPCR was performed with SYBR Green qPCR Master Mix on an ABI 7300 system. The relative mRNA expression levels were calculated using the 2−ΔΔCT method and normalized to GAPDH as an internal reference gene. Primer sequences, designed with Primer3 Input (version 0.4.0), are listed in Supplemental Table 3.
Statistical analysis
Statistical analysis was performed using the Mann–Whitney test to compare gene expression differences among the variants. Data were analyzed using SPSS version 23.0 (SPSS, Chicago, IL, USA), with a p-value < 0.05 considered statistically significant.
Results
Subcellular localization of variants
Similar to wild-type FOXL2, the proteins encoded by the double missense variants c.150C>G (p. Asp50Glu) and c.326A>T (p. Asn109Ile) were diffusely localized in the nucleus. In contrast, the proteins expressed from the deletion variants c.411_412del (p. Met137Ilefs101), c.533_542del (p. Val178Alafs90), and c.684delA (p. Ala229Leufs43) were also nuclear but aggregated. The proteins encoded by the nonsense variant c.214G>T (p. Glu72) and the duplication variant c.663_692dup (p. Ala225_Ala234dup) were retained in the cytoplasm (Figure 2).

Subcellular localization of wild-type and mutant FOXL2 proteins. Subcellular localization of FOXL2 as a fusion protein with EGFP (left). Nuclear stain with DAPI (middle). Merged image of left and middle (right). Both the wide-type FOXL2 and the double missense variation (p. Asp50Glu and p. Asn109Ile) diffusely show nucleus localization. Three indel variants of p. Met137Ilefs*101, p. Val178Alafs*90, and p. Ala229Leufs*43 are displayed nuclear aggregation (yellow arrows). The nonsense mutants of p. Glu72* and the duplication of p. Ala225_Ala234dup indicate cytoplasmic retention (red arrows). Scale bar: 5 μM.
Luciferase assay analysis
The transfected wild-type FOXL2 strongly inhibited the StAR promoter response compared to the negative control. In contrast, the mutant FOXL2 variants exhibited reduced inhibitory effects on StAR, particularly the c.214G>T (p. Glu72*), c.684delA (p. Ala229Leufs*43), and c.663_692dup (p. Ala225_Ala234dup) variants, which showed a significant decrease in inhibitory activity (p < 0.05) (Figure 3).
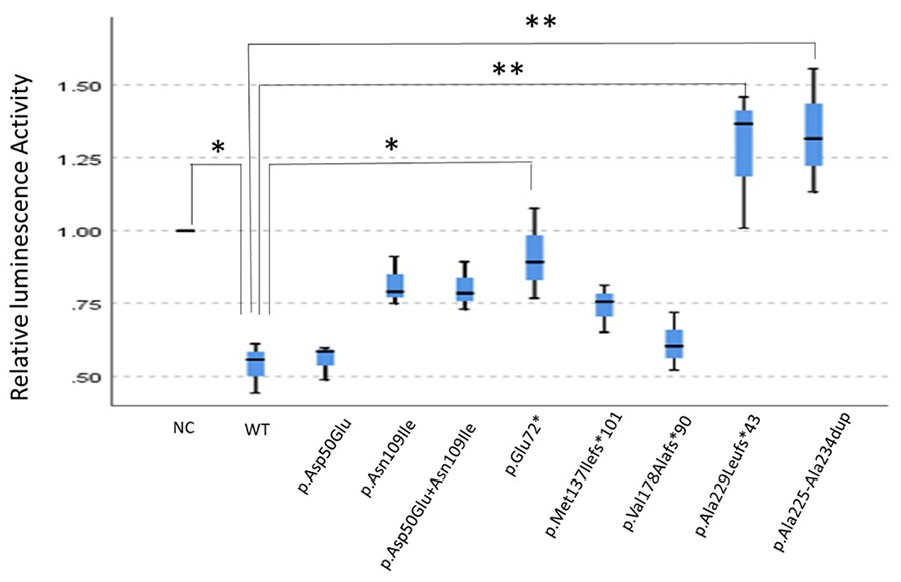
Inhibitory effects of different mutant types. All FOXL2 mutants show a reduced inhibitory effect on the StAR promotor response as compared to FOXL2-WT inhibitation, in which p. Glu72*, p. Ala229Leufs*43, and p. Ala225_Ala234dup indicate a statistically significant decreased inhibitory effect (p < 0.05). Types of different variants are shown on the X-axis, including negative control (NC) and wild-type FOXL2 (FOXL2-WT). Corresponding relative luminescence activity is shown on the Y-axis. Box plot displays the mean and distribution and skewness of the RLU data in quartiles. The blue box indicates the range of the median, with a horizontal line inside indicating the median and the upper and lower lines located outside the box representing the maximum and minimum of the data, respectively.
Expression level of FOXL2 downstream genes
In addition to its impact on StAR expression, mutant FOXL2 proteins also alter the expression of two downstream genes, CCND2 and INHBB. Similar to their effect on StAR, the mutant proteins reduce their inhibitory effect on the expression of both genes. Furthermore, the type of mutation correlates with the regulatory impact of the protein. Mutant proteins from nonsense and deletion variants exert a stronger influence on the expression of all three downstream genes, whereas those from missense variants do not (Supplemental Table 4).
Discussion
FOXL2 regulates transcription in the nucleus by binding to target genes through its FHD. Therefore, proper intracellular localization is essential for its function. A peptide segment, MFEKGNYRRRRRMK (codons 137–150), located at the C-terminal of the FHD, is believed to serve as the nuclear localization signal for FOXL2. Although the c.533_542del (p. Val178Alafs90) and c.684delA (p. Ala229Leufs43) variants produce truncated proteins, the nuclear localization signal remains intact in the abnormal peptides due to the preserved FHD. Similarly, the missense variants c.150C>G (p. Asp50Glu) and c.326A>T (p. Asn109Ile) do not disrupt the localization signal despite conformational changes in the protein, as the structural integrity of the FHD is maintained. Consequently, these aberrantly expressed proteins are still able to enter the nucleus.
However, the form of these abnormal proteins within the nucleus differs. Truncated proteins aggregate in the nucleus, potentially masking the FHD’s binding sites and disrupting FOXL2’s transcriptional regulation. In contrast, the conformational changes induced by missense mutations likely result in the loss of binding sites for target genes. Interestingly, the truncated protein produced by the c.411_412del (p. Met137Ilefs*101) variant was localized in the nucleus, despite the deletion starting at codon 137. We analyzed the structure of this truncated protein and identified a poly-arginine segment (RRRRRRRRVRR) predicted to span codons 169–177. We hypothesize that this poly-arginine fragment acts as a new nuclear localization signal, enabling the mutant protein to enter the nucleus. However, further studies are required to confirm this hypothesis.
In contrast, the truncated protein produced by the c.214G>T (p. Glu72*) nonsense variant lacks the FHD, including the nuclear localization signal, which results in the retention of the mutant protein in the cytoplasm. The c.663_692dup (p. Ala225_Ala234dup) duplication leads to an expansion of a 14-residue wild-type poly-alanine tract to 24 residues in the mutant form. This expansion is predicted to increase the volume of the mutant protein, hindering its nuclear entry. Additionally, the conformational changes may obscure the nuclear localization signal, further preventing its transport into the nucleus.
StAR is a key protein in the rate-limiting step of steroidogenesis and serves as a marker for identifying BPES types.8,10 Several genes contribute to ovarian granulosa cell development and germ cell proliferation, 11 including all genes selected for this study. However, only three genes—StAR, CCND2, and INHBB—were affected by the mutated protein in this study. These genes showed a significant reduction in gene expression inhibition by the mutated protein, particularly CCND2 and INHBB. This finding suggests that FOXL2 may regulate follicular development through additional pathways, such as those involving CCND2 and INHBB, in addition to the FOXL2-StAR pathway. Therefore, further research is needed to explore the impact of FOXL2 on other downstream genes.
FOXL2 functions as a transcriptional regulator within the nucleus. Genetic mutations that alter the location, structure, or morphology of FOXL2 in cells can impair its function, potentially influencing the disease morphology of FOXL2 in cells. In theory, a decrease in the amount of FOXL2 entering the nucleus correlates with a more significant disruption of transcriptional regulation. Reported FOXL2 mutations include missense, nonsense, deletion, and repeat mutations. However, since FOXL2 is a single exon gene, abnormal mRNA from nonsense and other frameshift mutations is expected to bypass the NMD surveillance mechanism. As a result, these abnormal mRNAs may enter the nucleus in altered forms or in reduced quantities, similar to the behavior of mRNAs from missense mutations. Abnormal proteins that enter the nucleus can cause disease through either loss of function or dominant negative effects. When abnormal mRNA does not enter the nucleus, the resulting disease is typically due to a loss of function.
This study aims to investigate the effects of various FOXL2 mutant types on nuclear localization and explore their impact on potential downstream target genes. However, there are several limitations to this study. First, HeLa cells, chosen as the cell model, are not ideal for studying granulosa cells or eyelid development, making it difficult to fully explain the pathogenesis of BPES through cell localization experiments. Second, we used transient gene expression to observe the effects of FOXL2 on downstream genes; however, stable gene expression would provide more reliable results. Establishing stable gene expression, though, would be time-consuming, especially given that the full range of downstream targets has not yet been identified. Finally, the potential impact of upregulated genes on FOXL2 expression remains unexplored, which represents another limitation of this study. In our experiments, we focus more on the effect of these mutations on their role in regulating gene expression, rather than exploring the mechanisms responsible for POF and BPES. As a regulatory factor, the ability to enter and exit the nucleus is crucial to its function. Therefore, we are more concerned with the effect of these variants on the incoming and outgoing nuclei.
FOXL2 mutations can cause blepharophimosis, often accompanied by POF. For female children, the effects of POF may be more significant than the cosmetic implications of blepharophimosis. However, current studies have not yet established a precise genotype–phenotype correlation, making early diagnosis and timely intervention for POF difficult. Our findings suggest that mutations in different regions or structural alterations may lead to varying degrees of dysfunction, providing a preliminary basis for establishing a relationship between genotype and phenotype.
Conclusions
In this study, we evaluate the functional changes induced by six pathogenic FOXL2 variants. We find that although the mutant proteins localize differently within the cells, all variants exhibit an inhibitory effect on gene expression. This inhibition is likely the primary cause of abnormal eyelid and ovarian development. Additionally, the double mutation did not result in a compounded effect on gene expression. The mutant protein may influence ovarian development through multiple pathways.
Supplemental Material
sj-docx-1-smo-10.1177_20503121251329287 – Supplemental material for Functional analysis of 6 variations in FOXL2
Supplemental material, sj-docx-1-smo-10.1177_20503121251329287 for Functional analysis of 6 variations in FOXL2 by Yuan Wang, Qian Wu, Yunyu Zhou, Wen Liu, Wenhong Cao, Yunwei Fan and Ningdong Li in SAGE Open Medicine
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.