
Abstract
Lipase maturation factor 1 is an endoplasmic reticulum-resident transmembrane protein, which acts as a critical chaperone necessary for the folding, dimerisation, and secretion of lipases. In this review, we summarise data about the recently revealed role of lipase maturation factor 1 in endoplasmic reticulum redox homeostasis, its novel interaction partners among oxidoreductases and lectin chaperones, and the identification of fibronectin and the low-density lipoprotein receptor as novel non-lipase client proteins of lipase maturation factor 1. Additionally, the role of lipase maturation factor 1-derived circular RNA in atherosclerosis progression via the miR-125a-3p/vascular endothelial growth factor A\Fibroblast Growth Factor 1 axis is discussed. Finally, we focus on the causative role of lipase maturation factor 1 variants in the development of hypertriglyceridaemia – a type of dyslipidaemia that significantly contributes to the development of atherosclerosis and other cardiovascular diseases via different mechanisms.
Keywords
Introduction
Hypertriglyceridaemia (HTG) is one of the most common types of dyslipidaemia affecting about 10% of the adult population worldwide and defined as having high plasma triglyceride (TG) levels. 1 While there are no globally defined threshold values to classify HTG, both fasting and non-fasting TG levels are usually applied. 2 Patients with HTG are at higher risk of atherosclerosis and other cardiovascular diseases (CVD)3–5 and pancreatitis. 6 HTG is often concomitant with other diseases, such as metabolic syndrome (MetS), type 2 diabetes (T2D), and obesity, thus contributing to their associated morbidity and mortality.7,8 HTG can manifest in various disorders, including familial chylomicronaemia syndrome (FCS), multifactorial chylomicronaemia syndrome, syndromic HTG, and autoimmune hyperchylomicronaemia, type III dysbetalipoproteinaemia, and others. 9
Mechanically, HTG affects the biosynthesis, metabolism, or clearance of TG, thereby increasing TG levels in the blood. Chylomicrons deliver TGs absorbed from the diet to skeletal and cardiac muscle, adipose tissue, and other tissues where lipases – lipoprotein lipase (LPL), hepatic lipase (HL), and endothelial lipase (EL) – break down the TG. 10 In general, 80% of FCS cases are caused by a mutation in the LPL gene, while the remaining cases are caused by variants in genes involved in LPL function and maturation – Apolipoprotein A-V, Apolipoprotein C-II, glycosylphosphatidylinositol anchored high-density lipoprotein binding protein 1 (GPIHBP1), or lipase maturation factor 1 (LMF1) – thus also affecting TG levels. 11 LMF1 is an ER membrane-localised chaperone protein consisting of an evolutionarily conserved LMF domain (PF06762 or IPR009613), which comprises five transmembrane segments that partition the protein into six distinct parts, with an N-terminal tail, loops B and D oriented towards the cytoplasm, and a C-terminal tail and loops A and C oriented towards the ER lumen. 12
The connection between HTG and the risk of CVD has been debated for many years. Thus, earlier meta-analyses demonstrated an absence of significant association between HTG and CVD risk, 13 while more recent meta-analyses and clinical studies have confirmed such association.14–17 Nonetheless, recent observational and interventional studies provided necessary support for the hypothesis that HTG is indeed a causal risk factor for CVD. In particular, patients treated with statins to maintain low-density lipoprotein cholesterol (LDL-C) levels but with elevated TG levels still exhibit a higher risk of CVD compared to those with effectively managed TG levels. 18 Furthermore, several clinical investigations have demonstrated that HTG remains independently associated with a higher CVD risk even after accounting for other lipid metabolism-related factors.19,20 At the same time, large-scale meta-analyses and clinical studies have confirmed that both fasting and non-fasting serum TG levels can serve as independent predictors of CVD risk.21,22
Further in this review, we focus on the recent findings deciphering the role of LMF1 in ER homeostasis, protein secretion, and maturation. Additionally, we describe the known molecular mechanisms regulating LMF1 expression and the role of LMF1-derived circular RNA in atherosclerosis. Finally, we discuss the recently identified LMF1 variants associated with HTG and the molecular mechanisms connecting HTG and atherosclerosis. PubMed, ScienceDirect, OpenMD, and Google Scholar search engines were used to search relevant recent publications and ‘LMF1’, ‘lipase’, ‘lipoprotein lipase’, ‘hypertriglyceridaemia’, and ‘lipase maturation factor’ were used as search word/s.
The functions of LMF1
The role in the post-translational maturation of lipases
Originally identified in 1983 as mouse mutation associated with combined lipase deficiency (cld), 23 this mutation was later named LMF1. 24 LMF1 is ubiquitously expressed in adult mouse tissues, and heterozygous mice displayed no notable abnormalities, while homozygous neonates suffered from severe HTG and perished within a few days after birth, primarily because of the near absence of enzyme activity for all three homodimeric lipases: LPL, HL, and EL. However, the monomeric pancreatic lipase was not affected. Interestingly, despite the mutation, the homodimeric lipases’ mRNA and protein levels remained normal in cld mice tissues, suggesting that the mutation-affected LMF1 chaperone function involved in the post-translational acquisition of lipases enzymatic activity.24,25 Similarly, a rare nonsense mutation (Y439X) was identified in the human LMF1 gene, and a patient homozygous for the Y439X mutation exhibited combined lipase deficiency accompanied by severe HTG, recurrent episodes of pancreatitis, tuberous xanthomas, and acquired partial lipodystrophy alongside T2D. 24 Later, a patient with another homozygous nonsense mutation (c.1395G > A, W464X) was reported and characterised by HTG, pancreatitis, and combined lipase deficiency, while tuberous xanthomas and lipodystrophy were not observed. The Y439X mutation completely eliminates LMF1 activity, whereas W464X represents a hypomorphic mutant, which reduces LPL activity and mass by 76% and 50%, respectively. 26
As shown in the LMF1-null mice model, LMF1 is ubiquitous and highly expressed in the normal mouse embryo; however, it is not essential for embryonic viability. Despite the absence of apparent morphological defects, the plasma LMF1−/− pups exhibited a milky appearance due to severe HTG (TG levels were ~80-fold elevated compared to controls) resulting from combined lipases deficiency. Additionally, total cholesterol (TC) levels were elevated, while HDL-C levels were not affected. Initially, the LMF1−/− genotype was represented at a Mendelian ratio (25%), but no LMF1−/− pups survived beyond the fourth day after birth. 24 Experiments on tissue-specific LMF1 over-expressing mice demonstrated that LMF1 expression was associated with increased LPL activity without changes in LPL mass. Furthermore, it was suggested that natural genetic variation in LMF1 (single-nucleotide polymorphisms – SNPs) may affect its expression and/or activity, thereby regulating lipase activity. 27
While early work on natural LMF1 mutants (Y439X and W464X) showed that the C-terminal tail is essential for proper lipase maturation,24,26 the functional role of other parts of the LMF1 protein remained unknown. Further research on LMF1 with truncated N-terminal region demonstrated that the full-length protein is necessary for LPL maturation. These truncated variants were correctly localised and oriented in the ER membrane; however, they were unable to rescue cld/cld cells. Moreover, evaluation of the endogenous level of LMF1 expression and LPL molecules showed that although LMF1 was expressed at a rather low level, each LMF1 molecule promoted the maturation of approximately 50 LPL molecules. 12
Additionally, because several naturally occurring isoforms of LMF1 and all isoforms of LMF2 28 have truncated LMF domain (PF06762), it has been proposed that LMF1 may play a broader role in ER homeostasis beyond its established function in the post-translational maturation of lipases.28,29
The role in ER-homeostasis
Three crucial ER quality-control mechanisms – ER-associated degradation (ERAD), unfolded protein response (UPR), and ER-phagy – are known to maintain ER homeostasis and adapt ER capacity in response to environmental signals. 30 ERAD targets misfolded secretory and membrane proteins for degradation by the proteasome, while autophagy can be engaged to clear protein aggregated within the ER. 31 Insufficient degradation of misfolded proteins in the ER leads to ER stress and activation of the UPR, which initiates widespread changes in transcription and translation. 32 The Hrd3-Hrd1 (hydroxymethylglutaryl reductase degradation protein) complex plays a central role in ERAD by targeting a specific subset of misfolded or unfolded proteins for proteasomal degradation. 33 The loss of Sel1L (Sel-1 suppressor of lin-12-like protein), the mammalian homologue of Hrd3, results in embryonic lethality, underscoring its significance in mammalian development. 34 Functionally, Hrd1 regulates Sel1L stability, and Sel1L is indispensable for maintaining ER homeostasis and ERAD in adult mice.35,36 However, the specific functions of Sel1L and ERAD in different cell types remain largely unexplored.
Interestingly, the mice with adipocyte-specific Sel1L deficiency (limited to white adipose tissue and brown adipose tissue) were resistant to diet-induced obesity but exhibited postprandial HTG and enlarged livers with steatosis. In mutant mice, postprandial LPL activity and mass were lower in plasma, but higher in white adipose tissue, with only 10%–20% LPL secreted compared to control mice, suggesting that Sel1L is indispensable for LPL secretion in adipocytes. Furthermore, Sel1L was shown to physically interact with and stabilise the LPL-LMF1 maturation complex (Figure 1). In the Sel1L-deficient cells, LPL was retained in the ER and formed aggregates that were cleared by autophagy. These data demonstrate that Sel1L, in addition to its role in ERAD, is involved in LPL secretion and, subsequently, in regulating and systemic lipid metabolism. 37

A model for the regulation and function of LMF1. ER stress regulates LMF1 expression in ATF6-dependent way. 38 LMF1-derived circular RNA negatively regulates miR-125a, which subsequently negatively regulates its downstream direct targets VEFGA and FGF1 – crucial players in atherosclerosis development. 39 Lipases (LPL, HL, and EL), fibronectin, and LDLR are known clients of LMF1, while other chaperones and disulphide bonds processing enzymes (ERp44, ERp72, ERdj5, calnexin, UGGT1, and UGGT2) directly interact with LMF1 and facilitate lipase maturation. 40 Hrd1 regulates Sel1L stability, while Sel1L interacts with and stabilises the LMF1-LPL complex. Negative regulation (red), positive regulation (black), indirect effect through several steps (dotted), direct interactions (blue), and LMF1 chaperone clients (dotted green double arrows). ER stress upregulates LMF1 (magenta arrow); treatment with the VSMCs proliferation and migration regulator PDGF-BB upregulates circLMF1, VEGFA, and FGF1 (cyan arrow), while downregulating miR-125a-3p (orange arrow).
Furthermore, the application of ER stress inducer tunicamycin 41 upregulated LMF1 expression in vivo, and further experiments on mouse embryonic fibroblasts and liver revealed that this effect was activating transcription factor 6 (ATF6)-dependent (Figure 1). 38 ATF6 is one of the tree ER quality control mechanisms (the other two being inositol-requiring enzyme 1 and protein kinase R-like endoplasmic reticulum kinase), which primarily induces the expression of genes involved in ER homeostasis. 42 Interestingly, ATF6 was sufficient to induce LMF1 expression even in the absence of ER stress, while ATF6 deficiency abolished tunicamycin-induced LMF1 upregulation. In total, these results demonstrated that LMF1 is a crucial player in ER homeostasis, and its function is not limited to lipase maturation. 38
Recently, the interaction of LMF1 with several lectin chaperones and disulphide bonds processing enzymes has been demonstrated. Among them, calnexin, endoplasmic reticulum protein 44 and 72 (ERp44 and ERp72), Endoplasmic Reticulum DNA J Domain-Containing Protein 5 (ERdj5), and UDP-Glucose Glycoprotein Glucosyltransferase (UGGT1 and UGGT2) have been identified as LMF1’s most significant interaction partners in HEK cell cultures (Figure 1). 40 Calnexin is a well-known type I integral ER membrane protein consisting of a transmembrane helix and cytosol-oriented C-terminal domain and is responsible for the folding and maturation of ER-synthesised glycosylated proteins. 43 Co-expression of LPL with calnexin increased LPL-specific activity by about three-fold, confirming LPL as its target. 44 Interestingly, other members of the ER molecular chaperone network (disulphide isomerases ERp44, ERp72, and ERdj5 45 ) play different roles in LPL and PL maturation. The knockdown of ERp72 decreased secretion of both LPL and PL, while the effects of ERp44 and ERdj5 knockdown were more pronounced for LPL. 40 Similarly, knockdown of UGGT1 and UGGT2, which selectively re-glucosylate unfolded glycoproteins, 46 decreased LPL secretion more dramatically than that of PL. 40
Because identified chaperones have catalytically active thioredoxin (TRX) domains and TRX-like domains, it was suggested that the cytosolic electron donor TRX may also be involved in LPL secretion. Indeed, the application of both siRNA and TRX inhibitor TXNIP reduced LPL secretion, while PL secretion was not affected. Further analysis of LMF1 with introduced point mutations of conserved C residues (C145A, C188A, C231A, C237A, C288A, C322A, C445A, and C453A) demonstrated that these redox-active cysteines interacted with TRX and ERdj5, thus contributing to the maintenance of the redox state of the ER (Figure 2). Finally, analysis of proteins whose secretion was compromised in cld/cld and cld/wt cells showed that fibronectin and the low-density lipoprotein receptor (LDLR) are additional non-lipase client proteins of LMF1 (Figure 1). 40 Interestingly, the oligomeric state, presence of disulphide bonds, and N-linked glycans 47 make fibronectin more similar to LMF1’s known lipase substrates than LDLR, a large multi-domain protein with 30 disulphide bonds and five N-linked glycans. 48
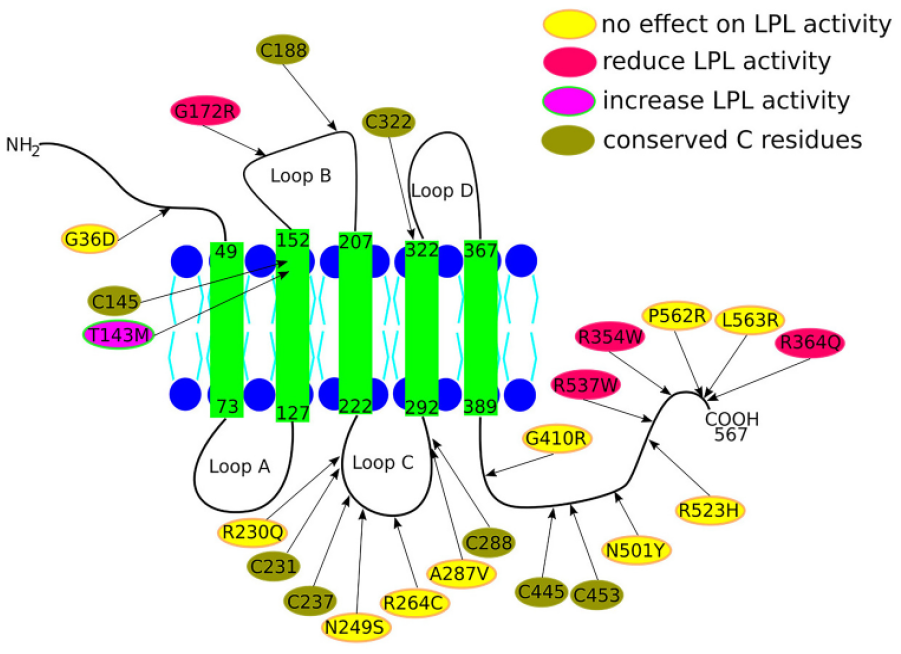
Schematic representation of LMF1’s topology, in vitro tested mutations, and conserved cysteine (C) residues.24–26,40 Mutations affecting lipases secretion/activity are depicted in red circles, mutations without significant effect on lipases are shown in yellow circles, and the only identified mutation that increases lipase activity, T143M, is depicted in magenta/green circle. Conserved C residues are shown in olive.
In total, these results demonstrate that LMF1 contributes to the regulation of ER redox homeostasis and the secretion of ER client proteins, which required the reduction of disulphide bonds during folding. Besides homodimeric lipases, fibronectin and LDLR have been identified as novel LMF1 client proteins. However, the exact mechanisms by which LMF1 performs these functions are not yet understood and require further investigation.
Role of LMF1-derived circRNA in atherosclerosis
In recent years, non-coding RNA has attracted significant attention as essential regulators and modulators of the progression of various diseases, including CVD. 49 A considerable amount of atherosclerosis research has focused on circular RNA (circRNA), a type of the non-coding RNA formed by back-splicing into covalently closed-loop structures, which makes them more stable and resistant to the RNA exonuclease-mediated degradation.50,51 Modulation of circRNA (e.g. circDHCR24, circ_0010283, and circRNA_0029589) levels has been successfully used to regulate vascular smooth muscle cell (VSMC) proliferation, viability, and migration in miRNA-dependent manner, thus confirming the important role of circRNA in atherosclerosis progression.52–54. Recent research has reported the role of LMF1 circRNA (derived from exon 6–8 of the LMF1 gene) in the development of atherosclerosis. 39 The levels of circLMF1, vascular endothelial growth factor A (VEGFA), and FGF1 were increased in platelet-derived growth factor-BB (PDGF-BB)-induced human aortic VSMCs, while the level of miR-125a-3p decreased (Figure 1). However, circLMF1 knockdown repressed cell viability, cell cycle progression, and migration of PDGF-BB-treated VSMCs. Mechanically, circLMF1 interacted directly with miR-125a-3p, which acted on its downstream targets VEGFA and FGF1. Indeed, miR-125a-3p upregulation resulted in reduced expression of VEGFA or FGF1. Overall, these findings revealed a novel role of LMF1-derived circRNA in atherosclerosis progression, at least partly through the miR-125a-3p/VEGFA\FGF1 axis, suggesting that circLMF1 can be a promising target for atherosclerosis treatment. 39
LMF1 variants associated with HTG
While mutations in the LPL gene are responsible for most reported cases of HTG, an increasing number of reports have identified novel mutations in other genes, including the APO family, GPIHBP1, and LMF1, that affect LPL-mediated TG-rich lipoprotein metabolism that can significantly elevate the risk of developing severe HTG.55–58 In the following section, we review recently discovered variants in the LMF1 gene in patients with HTG and related disorders (Table 1).
LMF1 variants identified in HTG patients.

CHZ: compound heterozygous; FCS: familial chylomicronemia syndrome; Het: heterozygous; HTG: hypertriglyceridaemia; HZ: homozygote; MA: mutant allele; MCM: multifactorial chylomicronaemia.
Unfortunately, the effect of the majority of identified LMF1 variants on lipases secretion/activity has not been tested under in vitro conditions, but has instead been predicted using different in silico tools. We have marked some in vitro tested variants on the LMF1 topology diagram (Figure 2). Interestingly, three out of four variants that decrease LPL secretion/activity (red circles) are localised on the C-terminal tail, while only one variant (G172R) is localised on the B loop. Neutral variants (with no significant effect on LPL – yellow circles) are localised on the loop C and the C-terminal tail (except G36D, which is localised on the N-terminal tail) (Figure 2). Furthermore, the T143M variant has been reported to increase LPL activity in vitro in one study, 69 but was characterised as neutral in another. 73 Thus, despite the fact that the selected variants tend to localise on the C loop and C-terminal tail, more studies are necessary to better understand the role of each structural element of LMF1 and the possible active sites in the process of lipases maturation and/or interaction with other co-chaperones and unknown bioactive molecules that can affect LMF1 functionality.
For readers, interested in other lipid metabolism genes, we wish to recommend recent papers discussing the role of Cholesteryl ester transfer protein and ATP-binding cassette transporter A1 (and their variants) in the development of CVD and associated diseases, design of novel diagnostic tools, therapeutic interventions, and treatment strategies.78,79
HTG and CVD risk
Atherosclerosis is a complex chronic inflammatory disease characterised by the pathological remodelling of arterial walls, resulting in their narrowing due to lipid accumulation and the formation of atheromatous plaques. The progression of atherosclerosis is linked to various medical conditions, including chronic kidney disease, peripheral artery disease, ischaemic stroke, and coronary artery disease. Collectively, CVD associated with advanced atherosclerosis contribute to 17.9 million deaths annually, accounting for 32% of all global deaths and making atherosclerosis the leading cause of mortality today. 80
In the initial stages of vascular remodelling, the innermost layer of blood vessels undergoes diffuse thickening, primarily driven by lipid cumulation and the recruitment of macrophages, which uptake multiple modified low-density lipoproteins (such as desialylation, oxidation, and other modifications). These macrophages are subsequently transformed into foam cells within the atherosclerotic plaque.81,82 The increased presence of macrophages, along with the release of inflammatory cytokines, the pro-apoptotic regulator Bcl-2-associated X protein, and transforming growth factor β, stimulates a transition of VSMCs towards a more fibroproliferative state, thus promoting the progression of the atherosclerotic lesion. In advanced stages, a thin fibrous cap forms, covering a necrotic lipid core that is often calcified. Such plaques are prone to rupture, leading to thrombus formation and vessel occlusion. 83 PDGF-BB is one of the crucial regulators of VSMCs proliferation and migration, as well as blood vessels formation and growth. 84
HTG contributes to atherosclerosis through several mechanisms. First, HTG increases the concentrations of triglyceride-rich lipoproteins (TGRLs) in the bloodstream, which exert a direct atherogenic effect. For example, TGRLs, such as apoB-containing lipoproteins, are sufficiently small to traverse the endothelium and possess significant pro-atherogenic potential on a per-particle basis. 85 Second, elevated plasma TG levels trigger various changes in the circulating lipoprotein profile, which are linked to an increased risk of atherogenesis. HTG enhances the activity of cholesteryl ester transfer protein, which facilitates the exchange of TGs for cholesterol esters (CE) between TG-rich and TG-poor lipoproteins. This process results in the cholesterol depletion of LDL and HDL particles, thereby reducing their particle size and cholesterol content. 86 The subsequent formation of small, dense LDL particles is more atherogenic than would be expected based on their cholesterol content alone, as there are multiple apoB molecules per unit of cholesterol. 85 Moreover, smaller cholesterol-depleted HDL particles are rapidly cleared by the kidneys, leading to decreased HDL-C levels and a reduced number of HDL particles. Even in patients with HTG who have LDL-C levels within the normal range, non-HDL-C (atherogenic cholesterol), and apoB (a marker of atherogenic particle count) levels are often elevated, thereby increasing the risk of atherosclerosis.87,88
Multiple lines of evidence indicate that elevated levels of TGRLs and remnant lipoprotein particles (RLPs) (such as chylomicrons and VLDL hydrolysed by LPL) are associated with a higher risk of CVD, even in individuals with LDL-C levels controlled by statins. 89 Another study demonstrated that individuals with high TG levels who were on statin therapy had a lower risk of death but a higher risk of CVD. 90 Moreover, recent Mendelian randomisation studies have provided causal evidence suggesting that lowering TG levels might increase the risk of thromboembolism but is likely to prevent Coronary artery disease and aortic valve stenosis. 91 Additionally, in statin-treated patients, non-HDL-C and apoB levels offer a more comprehensive reflection of the risk posed by atherogenic lipoproteins in both interventional and observational clinical trials compared to LDL-C. 92 Finally, current guidelines for managing HTG primarily emphasise lifestyle modifications, while statin therapy being considered based on an individual’s cardiovascular risk. 93
Recent evidence suggests that TGRLs and their derived RLPs, rather than TG or LDL-C alone, are the primary mediators of the increased CVD risk associated with HTG.94,95 Due to of the smaller size of RLPs compared to TGRLs, they can infiltrate the sub-endothelial space through transcytosis and contribute to lesion formation, 96 thereby linking postprandial HTG with an enhanced risk of CVD. 97 Specifically, intestinal chylomicron-derived RLPs, such as APOB48, have been positively associated with increased cardiometabolic risk in adolescents. 98
The role of HTG in foam cell formation and inflammation
Macrophages, which originate primarily from local proliferation of resident macrophages and the infiltration of circulating monocytes, play critical roles in the development of atherosclerosis. Once inside arterial walls, RLPs can be engulfed by lesional macrophages, leading to increased lipid accumulation within these cells and, subsequently, the formation of foam cells, a major hallmark of atherosclerosis. 99 The interactions between TGRLs/RLPs and macrophages may involve APOE on TGRLs/RLPs and the VLDL receptor on macrophages, potentially inducing these macrophages to adopt a pro-inflammatory M1-like phenotype. 100 Notably, very-low-density-lipoprotein receptor (VLDLR) associated with macrophages has been linked to foam cell formation and atherogenesis in various animal models and human cell cultures in vitro.101,102 Clinical studies have indicated that HTG and elevated RLP cholesterol are more causally related to low-grade inflammation than LDL cholesterol. 96 TGRLs, along with their LPL-mediated lipolytic products (such as RLPs and free fatty acids (FFAs)), increase the expression of adhesion molecules (Vascular cell adhesion molecule 1 and Intercellular Adhesion Molecule 1) and cytokines, thereby contributing to the development of atherosclerosis.103,104
Moreover, macrophage-derived LPL plays a role in foam cell formation and the development of atherosclerosis.105,106 In vitro exposure to FFAs induces TG accumulation and foam cell formation in macrophages. This is associated with the development of a pro-inflammatory phenotype and an increased uptake of modified LDL through the upregulation of the scavenger receptor CD36. 107 Additionally, TG synthesis within macrophages can drive inflammation by regulating prostaglandin E2. 108 TG accumulation in macrophages also upregulates the expression of multifunctional lactone hydrolysing enzyme paraoxonase 2 via the c-Jun N-terminal kinase signalling pathway, leading to inflammation and the generation of reactive oxygen species in the mitochondria.109,110 Furthermore, cholesterol and FFAs activate the NLR family pyrin domain containing three inflammasome in macrophages, which stimulates the synthesis and secretion of pro-inflammatory cytokines and promotes atherosclerosis. 111
Additionally, HTG is associated with increased lipids accumulation in circulating monocytes, leading to the formation of foamy monocytes in the circulation. 112 This lipid accumulation also promotes the production and priming of circulating pro-inflammatory monocyte-derived dendritic cells. 113 Furthermore, increased lipid levels within monocytes in individuals with HTG or MetS are linked to a shift in monocyte phenotype, characterised by elevated levels of adhesion molecules such as CD11b (Integrin alpha M) CD11c (Integrin alpha X), as well as cytokines like tumour necrosis factor alpha and Interleukin-1 beta, particularly in intermediate and non-classical monocytes.114,115 The severity of HTG and plasma TG levels is positively correlated with an increased number of total monocytes and M1 pro-inflammatory monocytes. 116 Consequently, lipid accumulation and phenotypic changes in circulating monocytes play a crucial role in the development and progression of HTG-associated CVD.
However, the mechanisms by which HTG and TGRL/RLP interact with circulating monocytes, leading to the formation of foamy monocytes, are not yet fully understood. While LPL-mediated lipolysis has been associated with HTG/TGRL-induced lipid accumulation and foam cell formation in tissue macrophages, recent reports suggest that HTG/TGRL can induce phenotypic changes even in LPL-deficient cells. 105 The VLDLR and LDL receptor-related protein-1 (LRP-1), both expressed by monocytes, may be involved in the interaction with and internalisation of TGRL/RLP, resulting in lipid accumulation in monocytes. 101 Additionally, APOE may play a significant role in HTG-mediated foamy monocytes formation and accelerate atherosclerosis. APOE is abundant in both TGRL and RLP, serves as a key ligand for both VLDLR and LRP-1, and plasma APOE levels are correlated with CVD mortality. 117
In total, HTG increases the risk of CVD through elevated levels of TGRL and their derivatives, such as RLP and FFAs. Beyond the direct effects of TGRL/RLP on ECs and lipid profile, these lipoproteins may also interact with and be engulfed by macrophages and monocytes. Consequently, inflammation in foam monocytes and macrophages, ECs adhesion, and lesion formation are likely to play crucial roles in the development of HTG-associated atherosclerosis.
Limitations and future directions
While the reviewed studies provide valuable insights, several limitations need to be addressed: (1) Incomplete understanding of mechanisms: The exact mechanisms by which LMF1 performs its functions are not yet fully understood. There is a need to elucidate how LMF1 regulates the maturation and secretion of client proteins, including its role in the reduction of disulphide bonds during folding. (2) Unknown role of LMF1 variants: The impact of various LMF1 gene variants on the activity of newly identified client proteins, such as fibronectin and the LDLR, remains unclear. Additionally, the connection of these variants to specific diseases has not yet been established. (3) Limited in vivo evidence: Although in vitro studies have provided insights into the effects of LMF1 variants, there is a lack of comprehensive in vivo data that could validate these findings and reveal how they translate to physiological conditions and disease states. (4) CircRNA functionality: While LMF1-derived circRNA has been identified as a novel player in atherosclerosis progression, its exact mechanisms of action through the miR-125a-3p/VEGFA/FGF1 pathway and potential interactions with other molecular pathways are not fully understood.
Based on pointed limitations, we wish to suggest several research directions for future exploration: (1) Mechanistic studies: Investigate the detailed mechanisms by which LMF1 facilitates the maturation and secretion of its client proteins. This includes understanding how LMF1 interacts with chaperones and disulphide bond processing enzymes, and its role in maintaining ER homeostasis. (2) Functional analysis of variants: Conduct studies to explore how different LMF1 variants affect the function of newly identified client proteins such as fibronectin and the LDLR. This could involve both in vitro and in vivo experiments to establish their relevance to disease states. (3) In vivo validation: Perform in vivo studies to confirm the findings from in vitro research. This includes using animal models to investigate how LMF1 variants and LMF1-derived circRNA influence physiological processes and contribute to disease progression. (4) CircRNA mechanisms: Elucidate the precise mechanisms through which LMF1-derived circRNA contributes to atherosclerosis. Research should focus on understanding its interactions with miR-125a-3p and its impact on VEGFA and FGF1, as well as exploring other potential molecular targets and pathways. (5) Therapeutic development: Explore the potential of targeting LMF1 and its derivatives (such as circRNA) in developing novel therapeutic strategies for atherosclerosis. This includes evaluating the feasibility and efficacy of such interventions in preclinical and clinical settings. (6) Broader client protein spectrum: Investigate other potential client proteins that interact with LMF1 and assess their roles in various diseases. This could help identify additional therapeutic targets and enhance our understanding of LMF1’s biological functions.
Conclusion
The LMF1 protein plays a crucial role in maintaining ER homeostasis, as well as in the maturation and secretion of ER client proteins, which requires the reduction of disulphide bonds during folding. This review discusses the current understanding of LMF1 regulation and function, its involvement in HTG, and its impact on atherosclerosis. Recent advances have identified fibronectin and the LDLR as novel client proteins of LMF1, alongside homodimeric lipases. Additionally, several lectin chaperones and disulphide bond-processing enzymes have been recognised as interaction partners of LMF1, which are essential for its proper functioning. LMF1-derived circRNA represents a completely novel factor in atherosclerosis progression, with its action through the miR-125a-3p/VEGFA\FGF1 pathway being the only mechanism identified to date. Furthermore, various mutations in the LMF1 gene have been clearly shown to affect lipase secretion and activity, linking them to HTG and CVD.
However, the exact mechanisms by which LMF1 performs its functions remain unclear and warrant further investigation. Additionally, the role of LMF1 variants in the activity of newly identified client proteins, such as fibronectin and the LDLR, is not yet understood, and no links to specific diseases have been established thus far. On the other hand, identification of an LMF1 variant that increases LPL activity in vitro, coupled with the discovery of LMF1-derived circRNA, suggests that LMF1 holds promise as a target for the development of novel therapeutic strategies for the prevention and treatment of atherosclerosis.
Footnotes
Author contributions
SAD and ANO conceptualised the manuscript; SAD wrote the manuscript text; AVC, VNS, DFB, INL, TIK, and ANO reviewed the text; AVC, DFB, and INL contributed to methodology; VNS, DFB, INL, and TIK contributed to formal analysis; VNS and ANO obtained funding and supervised. All authors have read and agreed to the published version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Russian Science Foundation, Grant# 24-15-00123 (conceptualisation; writing – original draft preparation; writing – review and editing; formal analysis; validation; funding acquisition; project administration).
Ethics approval
Not applicable.
Informed consent
Not applicable.
Trial registration
Not applicable.