
Abstract
Hereditary spastic paraplegia is a genetically heterogeneous neurodegenerative disorder characterised primarily by muscle stiffness in the lower limbs. Neurodegenerative disorders are conditions that result from cellular and metabolic abnormalities, many of which have strong genetic ties. While ageing is a known contributor to these changes, certain neurodegenerative disorders can manifest early in life, progressively affecting a person’s quality of life. Hereditary spastic paraplegia is one such condition that can appear in individuals of any age. In hereditary spastic paraplegia, a distinctive feature is the degeneration of long nerve fibres in the corticospinal tract of the lower limbs. This degeneration is linked to various cellular and metabolic processes, including mitochondrial dysfunction, remodelling of the endoplasmic reticulum membrane, autophagy, abnormal myelination processes and alterations in lipid metabolism. Additionally, hereditary spastic paraplegia affects processes like endosome membrane trafficking, oxidative stress and mitochondrial DNA polymorphisms. Disease-causing genetic loci and associated genes influence the progression and severity of hereditary spastic paraplegia, potentially affecting various cellular and metabolic functions. Although hereditary spastic paraplegia does not reduce a person’s lifespan, it significantly impairs their quality of life as they age, particularly with more severe symptoms. Regrettably, there are currently no treatments available to halt or reverse the pathological progression of hereditary spastic paraplegia. This review aims to explore the metabolic mechanisms underlying the pathophysiology of hereditary spastic paraplegia, emphasising the interactions of various genes identified in recent network studies. By comprehending these associations, targeted molecular therapies that address these biochemical processes can be developed to enhance treatment strategies for hereditary spastic paraplegia and guide clinical practice effectively.
Graphical abstract

Introduction
Hereditary spastic paraplegia (HSP) constitutes a diverse group of rare neurological disorders, typically arising from single-gene defects, with a global incidence rate of 3.6 per 100,000 individuals. 1 HSP is characterised by spasticity and paralysis in the lower limbs, resulting from developmental and neurodegenerative abnormalities in lower limb neurons. 2 Approximately 80 genes have been associated with HSP, including SPG3A, SPG4 and SPG11, which are prominent spastic paraplegia genes.2,3
Among the dominant HSP types (AD-HSP), SPG4 is identified as the most prevalent, accounting for 40%–45% of familial cases. 3 The second most common form of AD-HSP, SPG3A, is caused by mutations in the ATL1, estimated to contribute to about 7% of AD-HSP cases. 4 The autosomal recessive HSP type SPG11 is prevalent, affecting 18% of HSP patients globally, with the majority of cases found in Northern Africa and Western Asia. 1 Nevertheless, there is geographical variation in the prevalence of different HSP types, with SPG4 being the dominant form in German ethnic groups. 5
The pathogenesis of HSP involves a complex network of physiological processes, including membrane transport, endoplasmic reticulum (ER) shaping, mitochondrial functionality, DNA repair, autophagy, lipid metabolism and myelination. 1 Recent studies have also highlighted the potential roles of mitochondrial DNA (mtDNA) polymorphisms and abnormalities in endosome membrane trafficking and oxidative stress in the onset of the disease. HSP symptoms can manifest at any age, from infancy to late adulthood, with varying rates of progression and impairment.1,2 The most common genetic mutations and pathogenic pathways are illustrated in Figure 1.

Common genetic mutations and pathogenesis pathways in HSP. The diagram depicts the most common genetic mutations associated with HSP, as well as the pathogenesis pathways that contribute to the disease. The genetic loci for HSP are classified based on the mode of inheritance, and the figure highlights the common pathways that lead to HSP.
Clinically, HSP is categorised into two distinct groups: pure and complicated HSP, distinguished by the symptoms exhibited by patients. 6 Pure HSP typically presents with lower limb stiffness and urinary urgency, whereas complicated HSP is associated with a broader spectrum of symptoms, including signs of both upper and lower motor neuron dysfunction, such as fasciculations and increased spasticity. 1
In addition to primary neurological manifestations, HSP can lead to various secondary symptoms, encompassing paraesthesia, rigidity, scoliosis, malaise and muscle cramps and extending to cognitive dysfunction, ataxia and peripheral neuropathy. 1 The clinical picture is further complicated by the variability in symptom severity and age-related exacerbations that impact mobility and, consequently, the quality of life of affected individuals.
Recent advancements in artificial intelligence and machine learning have provided the means to utilise network analysis tools for the stratifying disease features in HSP, offering novel approaches to enhance our understanding of HSP subtypes. 7 By employing bioinformatics tools alongside molecular studies, this approach has revealed shared biological processes among clusters of HSP patients. Although further research is required to replicate and validate these findings, they suggest that patients can be grouped into distinct aetiological categories, based on molecular features that characterising HSP in each case. This implies that there may be more molecular interactions at play in the initiation and development of HSP than currently known. 7 By categorising patients with common molecular processes and comprehending all the mechanisms contributing to HSP development, this approach may pave the way for the development of precision medicine tools to improve patients’ quality of life.
Nevertheless, therapeutic options for HSP are presently limited, with no available disease-modifying interventions capable of reversing or slowing down disease progression. This review aims to explore the genetic basis of HSP, investigating the intricate relationships within interactome networks to unveil the metabolic mechanisms crucial to its pathophysiology. Understanding these associations may pave the way for targeted molecular therapy of various metabolic pathways, enabling the development of more effective HSP treatment algorithms and guiding valuable therapeutic strategies in clinical practice.
Methodology
This narrative review on the pathogenesis and management of HSP employed a rigorous methodology to ensure a comprehensive exploration of the topic. The inclusion criteria encompassed studies of various designs, including observational studies, case–control investigations, cohort studies and randomised controlled trials. Furthermore, the review considered studies involving both paediatric and adult populations, as well as those addressing all types of HSP. There was no set time frame for study inclusions, although we prioritised recently published papers to reflect on current advances on the topic. The literature search was conducted through prominent databases, including Scopus, MEDLINE, EMBASE and the Cochrane Library. The search was characterised by the use of precise search terms, such as ‘HSP’, ‘management’, ‘treatment’ and ‘pathogenesis’. Additionally, a manual search was performed to identify references from recently published reviews. It is important to note that stand-alone abstracts and unpublished studies were excluded from the review.
Through this comprehensive and methodical approach, the review aspires to offer a high-quality academic assessment, presenting novel insights into the pathogenesis and management of HSP. This will facilitate a thorough synthesis of pertinent findings and contribute to a deeper understanding of this complex condition. A summary of the methodology is provided in Table 1 for reference.
Summary of methodology for pathogenesis and management of HSP.

Comprehensive overview of HSP pathophysiology
Oxidative stress and mitochondrial dysfunction
Oxidative stress and mitochondrial dysfunction are central to the pathogenesis of HSP. Neurons have a high energy demand to maintain their resting membrane potential and support axonal transport, especially in long axons. Therefore, the proper functioning and organised distribution of mitochondria within neurons are crucial. 8
One key aetiological element in HSP pathogenesis is the absence of the paraplegin/AFG3L2 complex in the inner mitochondrial membrane. This complex not only prevents neuronal degeneration but also sustains the oxidative phosphorylation pathway, responsible for generating adenosine triphosphate (ATP). 9 In SPG7 patients with loss-of-function mutations in this complex, cells become more vulnerable to oxidative stress, leading to increased sensitivity to reactive oxygen species and reduced mitochondrial ATP production.9,10 This can further impair neuronal function and contribute to the onset of HSP.
Disruption of mitochondrial fission–fusion dynamics can lead to morphological, mobility, and spatial distribution changes in mitochondria. 8 This results in impaired mitochondrial axonal transport, leading to deficiencies in oxidative phosphorylation, apoptosis, deletion mutations in mtDNA, and ultimately axonal degeneration. 8 Several HSP subtypes, including SPG15, SPG48 and SPG31, have been linked to impaired mitochondrial dynamics.11,12 Meanwhile, SPG7 and SPG61 are associated with mitochondrial morphology and network disruptions. 13 Notably, the SPG7 gene encoding paraplegin, a protein found in the inner mitochondrial membrane, and the SPG13 gene encoding heat shock protein-60, a mitochondrial chaperone, are both related to mitochondrial dysfunction and spastic paraplegia. 14 Mutations in mitochondrial fusion–fission genes have also been reported in various HSP types, including dynamin-related protein 1 (DRP1), optic atrophy gene 1 and mitofusin-1 and -2 (Mfn1 and Mfn2).15,16
Mitophagy is another crucial process in the regulation of mitochondrial dynamics and function within axons. 17 Disruption of mitophagy can lead to the formation of abnormal mitochondrial networks and subsequent axonal degeneration. 17 HSP subtypes such as SPG11, SPG15 and SPG48 result from autosomal recessive mutations in the genes SPG11, ZFYVE26 and AP5Z1, encoding spatacsin, SPASTIZIN and AP5Z1, respectively. 18 These genes hold promise for the discovery of new therapeutic targets for these HSP subtypes. Recent research has shown that inhibiting the overexpression of DRP1, a mediator of mitochondrial fission, can reverse axonal defects observed in neurons of SPG15 and SPG48-HSP subtypes. 8
Finally, the accumulation of mitochondria within axons, lack of ATP and disruption of the neuronal cytoskeleton can result in axonal swelling, leading to various neurological disorders, including HSP.11,12
Metabolic processes influencing HSP
Disturbed ER function and HSP
Disrupted ER function is a significant contributor to the pathogenesis of HSP. The ER is essential for proper lipid synthesis and metabolism and facilitates intracellular communication by generating microdomains in plasma membranes. 19 Neurodegenerative processes have been linked to ER and lysosomal malfunction, where sphingolipids are produced and degraded. Sphingolipids are crucial for normal neuronal structure and functionality. 19 Patients with a missense mutation in fatty acid-2-hydroxylase, the enzyme responsible for sphingolipid production, are at risk of developing complex HSP. 20 Additionally, deficiencies in proteins like Receptor Expression-Enhancing Protein 1 (REEP1), which play a vital role in shaping the tubular ER network, can lead to shorter corticospinal axons and rapid declines in motor function tests, indicative of early onset HSP. 21
The spastin gene, typically involved in conventional microtubule production and cellular stability, has a strong association with HSP development. 22 Recent research has shown that spastin dysfunction leads to the accumulation of lipid droplets (LDs) within neuronal cells. 22 While more research is needed to fully understand these novel findings, it is plausible to hypothesise that HSP pathogenesis may involve an alternative mechanism of abnormal LD accumulation and metabolism in addition to the well-known spastin gene dysfunction. 23
Moreover, three distinct mutations – frameshift, whole-gene deletion and single missense mutations – within the reticulon 2 (RTN2) gene have been associated with aberrant ER morphogenesis in SPG12. 24 RTN2 encodes a member of the reticulon family, which serves as prototypic ER-shaping proteins in families afflicted by SPG12. The truncated RTN2 protein is believed to act through a haploinsufficiency mechanism, although the possibility of a toxic gain of function in the cytosol or nucleus remains. 24 The interaction between RTN2 and spastin is contingent on a hydrophobic region within spastin, a region involved in ER localisation and projected to form a curvature-inducing/sensing hairpin loop domain. 24 These findings collectively suggest the involvement of a reticulon protein in axonopathy as part of a network of interactions among HSP proteins engaged in ER shaping. This supports the prevailing hypothesis that ER dysfunction constitutes a pathogenic mechanism in HSP.
Ornithine metabolism and HSP
The ornithine and HSP genes are illustrative instances of metabolic pathways that exhibit multiple links with lipid metabolism. 25 Ornithine, a non-essential amino acid found in the urea cycle, is recognised for its pivotal role in various metabolic processes. Dysregulation of ornithine metabolism has been associated with several diseases, including leukaemia, hyperammonemia, neuroblastoma and neurodegenerative syndromes like HSP. 25
Mutations in the ALDH18A1 gene, which participates in the ornithine pathway, have been correlated with the pathogenesis of HSP. 25 The ALDH18A1 gene encodes the P5CS enzyme, which catalyses the initial step in the biosynthesis of proline and ornithine.25,26 This enzyme converts gamma-glutamyl phosphate into gamma-glutamyl semialdehyde, which subsequently undergoes spontaneous degradation to P5C. Located in the inner mitochondrial membrane, P5C can then be further metabolised into proline or, through the urea cycle, into ornithine, citrulline and ultimately arginine, with the assistance of the P5C reductase (PYCR1) enzyme (Figure 2).25,26 Consequently, low levels of plasma ornithine are a matter of concern for individuals with HSP.

The ornithine metabolic cycle and its association with lipid metabolism in relation to HSP.
HSP resulting from ALDH18A1 gene mutations can manifest as either a simple autosomal recessive or complex autosomal dominant condition. Patients from autosomal dominant families consistently exhibit low to extremely low plasma citrulline levels, along with less consistently reduced plasma levels of ornithine, arginine and proline, regardless of the specific protein domain affected by the mutation. 25 Notably, patients with autosomal dominant HSP linked to ALDH18A1 mutations may derive substantial benefits from long-term citrulline supplementation, a therapeutic approach that has demonstrated success in patients with inborn metabolic disorders affecting the urea cycle, either directly or indirectly, such as ornithine transcarbamylase deficiency or lysinuric protein intolerance.
Lesser known mechanisms in HSP
Spastic paralysis is a frequently observed phenotype in various rare mutations affecting interconnected pathways related to glutamate and urea cycle metabolism, as well as mitochondrial degeneration. 27 Notable examples of such mutations involve PYCR1 and NMNAT1 genes, which are associated with cutis laxa and the nicotinamide adenine dinucleotide (NAD) biosynthetic network, respectively. 27 A comprehensive examination of the underlying pathology associated with these mutations can provide valuable insights into our comprehension of HSP (Figure 3).

Lesser known mechanisms in HSP.
PYCR1 mutations
PYCR1 mutations have been implicated in an autosomal recessive neurocutaneous syndrome associated with a range of clinical manifestations, including mental retardation, cutis laxa, joint hyperlaxity, progeroid dysmorphia, growth retardation, microcephaly and corpus callosum dysgenesis. These mutations are also believed to play a role in neurodegeneration within the mitochondria. 28
The PYCR1 gene encodes the enzyme P5C reductase, which is responsible for the final step in proline biosynthesis, converting P5C into L-proline. 28 Notably, although proline plasma levels remain normal in individuals with PYCR1 mutations, they exhibit mitochondrial abnormalities characterised by increased apoptosis during oxidative stress, resulting in the development of cutis laxa with progeroid changes. 28
It is worth noting that mutations in the PYCR1 gene, as well as P5CS deficiency associated with the ornithine cycle, share common features, including connective tissue defects and developmental delays. Both enzymes are situated in the inner membrane of mitochondria.27,28
NMNAT1 mutations
The Nicotinamide mononucleotide adenylyltransferase enzyme plays a crucial role as a co-substrate for a group of enzymes within the NAD biosynthetic network. Recent evidence from whole-exome sequencing indicates a potential link between a variant in NMNAT1 and HSP. 29 This association was observed in two affected siblings diagnosed with HSP, where a homozygous variant (Glu257Lys) in NMNAT1, the most prevalent variant in patients with LCA9, displayed clinical manifestations completely consistent with pure HSP. 29 Notably, this study represents the first report of NMNAT1 mutations contributing to neurological disorders by disrupting the regulation of physiological NAD homeostasis. To validate this hypothesis, further rigorous and large-scale clinical trials are imperative.
Cellular transport mechanisms and HSP
Malfunction in axonal transport represent an additional pathophysiological process associated with specific subtypes of HSP. 12 Axonal transport plays a vital role in various essential functions, encompassing the regulation of axon composition, the preservation of axonal function, and the support of neurogenesis. 12 This process relies on motor proteins, including kinesin for anterograde transport, which facilitates the movement of ribonucleic acids (RNAs), proteins, lipids and mitochondria towards axon terminals and growth cones. In contrast, retrograde transport is orchestrated by dynein motor proteins, responsible for transporting trophic factors and misfolded proteins back to the soma for degradation. 30
Kinesin heavy chain 5A (KHC5A) is a pivotal anterograde motor protein belonging to the kinesin family that is responsible for transporting neurofilaments and vesicles to growth cones and synapses. 31 It also plays a regulatory role in cargo transport within dendrites and is involved in processes such as exocytosis and endocytosis. The KHC5A protein is encoded by the KIF5A gene, which has been found to be mutated not only in SPG10 but also in other neurodegenerative diseases, including Charcot–Marie–Tooth (CMT) disease Type 2 and amyotrophic lateral sclerosis (ALS). 31 These mutations predominantly manifest as missense changes and tend to cluster in the gene’s N-terminal motor domain, as indicated by comprehensive genome-wide analyses. A similar anterograde protein, the Kinesin Family Member 1A protein (KIF1A), encoded by the KIF1A gene, is responsible for the transport of membranous organelles along the axon. 31 Notably, Klebe et al. 32 employed targeted next-generation sequencing to establish that mutations in the KIF1A gene underlie the development of SPG30.
KIF1A gene mutations have also been associated with other neurological conditions, including hereditary sensory neuropathy type IIC and nonsyndromic intellectual disability. 33 On the other hand, the SPAST gene, mutated in the most prevalent form of autosomal dominant HSP, SPG4, encodes for spastin, a protein pivotal in regulating microtubule dynamics and arrays.34,35 As the normal functioning of kinesin and dynein motor proteins relies heavily on microtubule activity, any dysregulation stemming from mutant spastin proteins can lead to defective axonal transport. 36 Another analogous protein, Spartin, encoded by the SPART gene, is also associated with microtubule dynamics, in addition to its involvement in endosomal trafficking, and has been implicated in SPG20, also known as Troyer syndrome. 37
Distinctively, SPG33 is a subtype of HSP resulting from mutations in the ZFYVE27 gene, which encodes for protrudin. Protrudin is a protein localised to the ER and plays a role in the outgrowth of neurites. 18 SPG33 stands out as its causative protein, protrudin, interacts with proteins associated with several other spastic paraplegias. It forms connections with both SPG4 and atlastin-1 (SPG3A), while also interacting with the KIF5 proteins (SPG10). 18
Membrane trafficking in ER networks
HSP-associated genes have been identified to play pivotal roles as modifiers and regulators of the ER network, impacting protein secretion and calcium sequestration. 38 The ER, a membrane-bound organelle, participates in a multitude of cellular processes and, most notably, plays a critical role in expanding axonal membranes to facilitate postsynaptic signalling pathways. 38 Mutations that disrupt ER shaping and function contribute not only to the phenotypic spectrum of HSP but also to various other neurodegenerative conditions, including Alzheimer’s disease (AD), CMT, Parkinson’s disease (PD) and ALS. 38
Genetically, HSP can be inherited in different modes, including autosomal dominant, autosomal recessive, X-linked or mitochondrial. However, the most prevalent form of HSP is autosomal dominant HSP, affecting 75%–80% of HSP patients. 39 Dysregulation in the tubular ER network can largely be attributed to three main genes: SPG3A/ATL1, SPG4/SPAST and SPG31/REEP1, accounting for approximately 50% of HSP cases. 40 Heterozygous mutations in the ATL1 gene result in SPG3A AD-HSP. ATL1 encodes the GTPase atlastin, which interacts with spastin. 41 Atlastin-1 is instrumental in shaping the ER morphology by catalysing the fusion of membrane tubules, forming three-way junctions. 42 Mutations in this gene culminate in the abnormal morphogenesis of the ER and Golgi, thereby impacting axon development and maintenance, ultimately contributing to HSP. 43 The SPAST gene encodes for spastin, which is part of the ATPases Associated with the diverse cellular Activities (AAA) family. Spastin plays a crucial role in mediating microtubule stability, and its loss has been shown to result in defective synaptic growth and neurotransmission in vivo in Drosophila.44,45 This highlights spastin as a potential therapeutic target for HSP treatment.
Mutations in the REEP1 gene are another well-recognised cause of SPG31 AD-HSP. 46 Previous studies yielded conflicting data regarding the localisation of REEP1, with both the ER and mitochondria being suggested. However, an in vitro study clarified that REEP1 is localised in the ER-mitochondria interface. Pathological REEP1 mutations disrupt ER-mitochondria interactions, leading to growth defects and neuronal degeneration, which can be attributed to the inability to maintain healthy long axons in HSP. 47 Within the ER, REEP1 forms protein complexes with both atlastin-1 and spastin, necessitating the presence of hydrophobic hairpin domains in these proteins. 47 Further in vitro research highlighted the role of REEP proteins, particularly those with extended C-terminal domains (REEP1–4), in ER shaping and their involvement in interactions between ER tubules and the microtubule cytoskeleton. 47 Notably, an SPG31 mutation in REEP1 without a C-terminal domain was found to fail to interact with the ER membrane in corticospinal neurons, potentially contributing to the pathogenesis of HSP. 47
The BSCL2 gene, also known as the Berardinelli–Seip congenital lipodystrophy gene, encodes the ER protein seipin. Mutant seipins in BSCL2-associated HSP have been found to accumulate in the ER, triggering the upregulation of ER-stress-mediated molecules, which, in turn, induce apoptosis. This mechanism provides insight into the degeneration observed in HSP. 48
The ER is co-localised by protrudin, a membrane protein involved in polarised vesicular trafficking in neurons. 49 In vitro studies demonstrated that the forced expression of protrudin promotes the formation and stabilisation of the ER tubular network. Conversely, mutant protrudin cells exhibited greater susceptibility to ER stress, potentially contributing to the pathogenesis of HSP. 49
Vesicular trafficking of lysosomes
Lysosomes are dynamic organelles enclosed by a single membrane, displaying significant heterogeneity in terms of size, location, shape, enzyme composition and substrates within cells. These organelles house a multitude of integral and peripheral membrane proteins, including transporters and ion channels.50 –52 The acidic environment of the lysosomal lumen, with a pH typically ranging from 4.5 to 5.5, is primarily maintained by the lysosomal multi-subunit V-ATPase. In this low-pH setting, over 50 intralysosomal hydrolases become activated, enabling the breakdown of macromolecules like proteins, nucleic acids, lipids and carbohydrates.52 –54
Lysosomes play a crucial role in importing and degrading various substances, including endocytic materials like small molecules and cell surface proteins, phagocytosis of larger particles such as inflammatory cells and exogenous bacteria, and autophagy of cytoplasmic components like damaged mitochondria, ER and other lysosomes.52 –54 These organelles act as cellular waste disposal sites, recycling the by-products of lysosomal digestion for cellular maintenance.
Lysosomal dysfunction is clinically implicated in various human disorders, including lysosomal storage diseases such as Tay–Sachs disease and neurodegenerative diseases like ALS and HSP.55,56 In HSP, genes like the kinesin family member 5A gene (KIF5A) and protrudin play essential roles in vesicular trafficking and ER-lysosome interactions, respectively, contributing to the pathogenesis of the disease.57,58 Additionally, lysosomal proteins such as VPS35 (vacuolar protein sorting 35 orthologs), a component of the Retromer complex that sorts endosomal products, have been associated with HSP. 59
The absence of components of the Wiskott–Aldrich syndrome protein and SCAR homologue 1 (WASH-1) complex, which interacts with endosomes, as well as the absence of FAM21 and actin-capping proteins responsible for regulating actin dynamics, can result in increased tubulation of early endosomes. This change may be due to the lack of actin-mediated forces needed for the separation of tubular transport intermediates from endosomes. This alteration affects the transport of early endosomal compartments and is particularly relevant in the case of corticospinal neurons, which have exceptionally long axons and are more susceptible to reductions in functional WASH. This susceptibility could be a contributing factor in HSP. 60
Furthermore, endosomal membrane trafficking in HSP is associated with specific proteins, including KIAA0415 (SPG48), spatacsin (SPG11) and spastizin/FYVE-CENT (SPG15). Clinical similarities between SPG11 and SPG15 are noted, including their presentation as juvenile parkinsonism and a connection with the thin corpus callosum. The interaction of spastizin and spatacsin with the SPG48 protein KIAA0415 suggests a link between HSP and DNA repair. 61
Macro-autophagy
Autophagy is a fundamental cellular process that enables cells to respond effectively to potential disruptions, safeguarding the cellular environment from organelles and misfolded proteins that could compromise internal balance. 62 Macro-autophagy, a central aspect of autophagy, involves the sequestration of cytoplasmic components into double-membrane vesicles known as autophagosomes, which subsequently merge with lysosomes to form autolysosomes. 62 This pivotal cellular process is indispensable for normal cell function, and any defects in autophagy have far-reaching implications, particularly in the context of neurodegenerative conditions like HSP. 63 The malfunction of the autophagic pathway, in conjunction with endolysosomal processes, has been associated with 24 gene mutations linked to HSP, signifying its pivotal role in the pathogenesis of HSP (Figure 4). 63
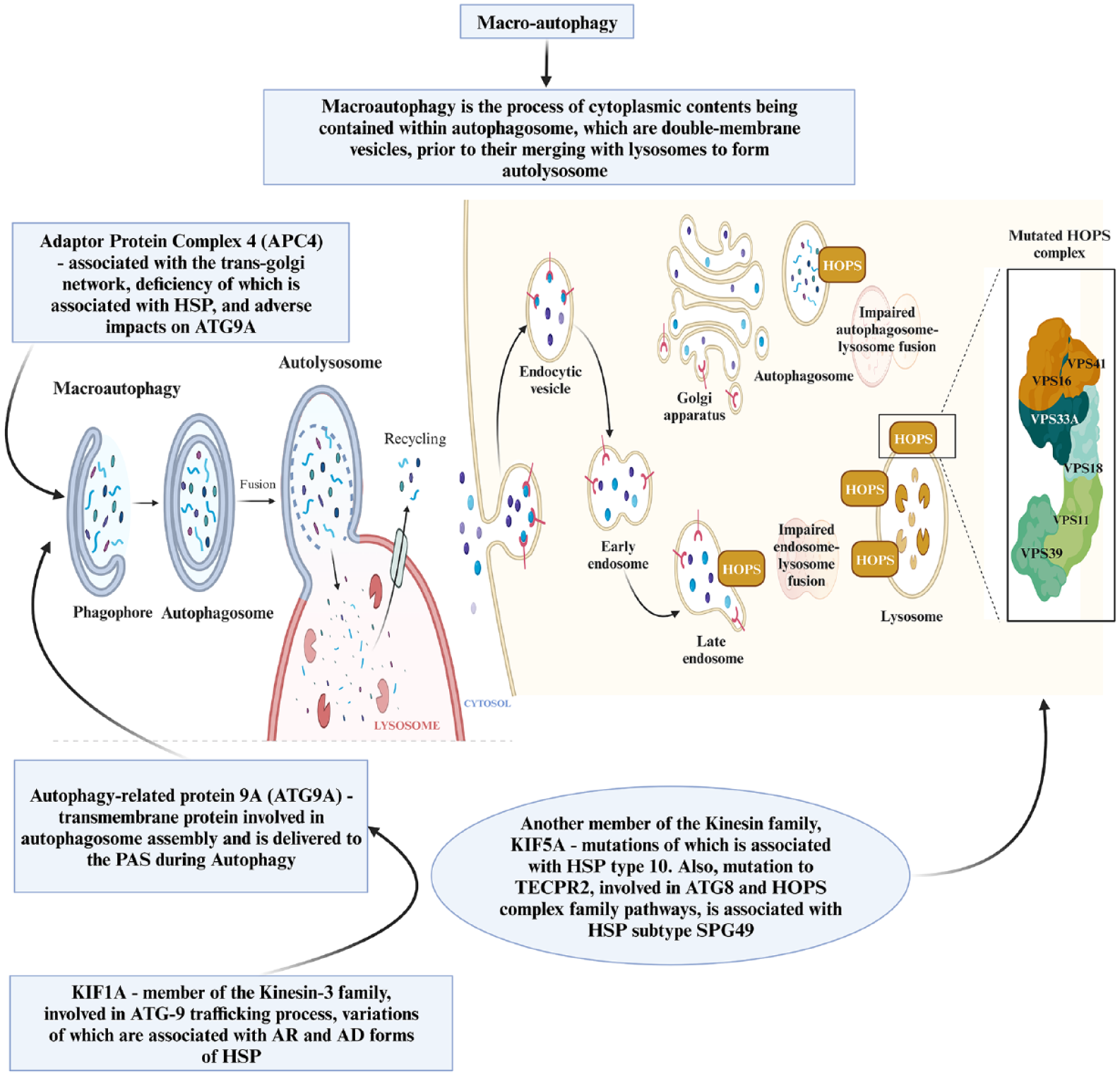
Macro-autophagy.
The autophagosome
In the process of autophagosome biogenesis, malfunctioning proteins serve as attractants for autophagy-related (ATG) proteins, initiating the creation of phagophore assembly site (PAS) and ultimately giving rise to the formation of a phagophore. 64 This sequence is then succeeded by nucleation and elongation phases, culminating in the generation of an autophagosome that encapsulates intracellular material.
Within this process, ATG protein 9A (ATG9A) plays a crucial role. ATG9A, a transmembrane protein, is indispensable for the assembly of autophagosomes. During autophagy, ATG9A is transported to the PAS by vesicle transport mechanisms and takes on the pivotal role of overseeing the regulation and elongation of the phagophore (Figure 4). 64
Adaptor protein deficiency syndromes, the Kinesin family and autosome-lysosome fusion in HSP
Adaptor Protein Complex 4 (AP-4) is a complex predominantly associated with the trans-Golgi network (TGN). Depletion of this complex has been linked to HSP and has direct consequences on ATG9A, including its mislocalisation and retention at the TGN rather than reaching the PAS). 65 Mutations in AP-4 have been identified in severe neurodevelopmental forms of HSP, specifically SPG47, SPG50, SPG51 and SPG52, collectively known as AP-4 deficiency syndromes. 66
Another pivotal player in the ATG9A trafficking process is KIF1A, a member of the Kinesin-3 family. ATG9A is typically located at the PAS and axon terminals. 32 Pathological variations in KIF1A have been associated with impaired transport of ATG-9 to neurites and are linked to both autosomal recessive and dominant forms of HSP, including SGP30. 32
Furthermore, several other genes in the Kinesin family have been implicated in HSP. Mutations in KIF5A, which encodes the heavy chain of kinesin-1, are known to disrupt axonal transport, particularly affecting neuronal microtubule motors. This disruption is associated with SPG10-HSP. 67 These kinesin motors play essential roles in the fusion of autophagosomes with lysosomes, and their differential intracellular locations necessitate trafficking. 67 Another protein involved in the union of autophagosomes and lysosomes is Light Chain 3 (LC3), a member of the ATG8 protein family, which recruits interacting partners to initiate transport, tethering, and fusion, including the Homotypic Fusion and Vacuole Protein Sorting Tethering Complex (HOPS). 68
Additionally, the Tectonin Beta-Propeller Repeat Containing 2 (TECPR2) protein is associated with pathways involving ATG8 and the HOPS complex family. Mutations in the TECPR2 gene have been linked to SPG49-HSP. A study examining fibroblasts from an HSP patient revealed defects in autophagy associated with TECPR2 mutations. 69 It is suggested that TECPR2 regulates autophagosome-lysosome tethering and fusion by interacting with LC3 and HOPS (Figure 4). 70
Impaired lysosomal storage and HSP
ATP13A2 is an enzyme closely associated with lysosomal transport and SPG78-HSP. This enzyme belongs to the P5-type transport ATPase family and plays a pivotal role in lysosomal function. Individuals with ATP13A2 mutations often exhibit an abnormal accumulation of lysosomal and autophagosome structures, resulting in impaired lysosomal storage. This disruption leads to a defect in the clearance of damaged mitochondria and hinders the degradation of intracellular protein aggregates. 70
Once autophagosomes have tethered to lysosomes to form autolysosomes, the internal materials contained within are subjected to degradation. The final step in autophagic pathways is the recycling of lysosomal components, which facilitates the formation of new lysosomes during reformation. 70
Multiple proteins linked to HSP are known to play roles in the process of lysosomal reformation, including spatacsin (SPG11), spastizin (SPG15), AP5Z1 (SPG48) and the AP-4 complex. AP5Z1 is a part of the adaptor protein complex 5 (AP-5) and is closely associated with both spatacsin and spastizin. 71 Mutations in AP5Z1 can result in the loss of AP-5 protein function, leading to abnormal storage of lysosomal materials (Figure 4). 72
Myelination disruptions and HSP
PLP1
The PLP1 gene encodes proteolipid protein 1. Mutations in the PLP1 gene itself or in its distal enhancers have been identified as contributing factors to HSP and specifically as causative factors for with SPG2 being one of the specific manifestations linked to these mutations.73 –75 Notably, a microdeletion at the Xq22.2 region, which contains one of the distal enhancers of the PLP1 gene, has been found to lead to SPG2-HSP. 73 When these distal enhancers are absent or impaired, the transcription of PLP1 in oligodendrocytes can be suppressed. This transcriptional suppression can limit the ability of the gene to maintain myelin, contributing to the development of the SPG2 phenotype. 73
L1CAM
The L1CAM gene is responsible for encoding myelin proteins, which play a crucial role as they are expressed on oligodendrocytes that provide vital support to motor neurons. 76 When mutations occur in the L1CAM gene, it results in a deficiency of myelin proteins in neurons. 76 Notably, in the axons of neurons expressing the Trk-fused gene (TFG) mutation (p.R106C), there is a reduction in the levels of cell surface L1CAM when compared to controls. This observation supports the idea that the transport of L1CAM to the cell surface is compromised in these conditions.77,78 These findings underscore the significance of effective membrane trafficking in the early secretory pathway, especially in the context of the long corticospinal motor neurons that degenerate in cases of HSPs. Such efficient trafficking is essential for maintaining neuronal homeostasis. 77
Intersecting genes and metabolic pathways in diseases related to HSP
Familial ALS
Familial ALS (fALS) is a rapidly progressing neurodegenerative disease that leads to paralysis. 79 Multiple genes, including BSCL2, ERLIN1, ERLIN2, SPG11 and more, are thought to contribute to fALS. Differentiating between HSP and fALS can be challenging as each of these genes interacts with the metabolic pathways associated with both conditions. 80
Seipin is a protein encoded by the BSCL2 gene. Seipin is highly prevalent in the brain, particularly in spinal motor neurons and cortical neurons in the frontal lobes. It is a membrane protein found in the ER of various cell types and is believed to be involved in the formation of LDs. 81 In fALS, this gene and its variants, such as P.N88S, P.S90L and P.S90W, 82 exhibit a harmful gain-of-function mutation. This mutation results in the misfolding of Seipin protein, leading to its accumulation in the ER, causing neurotoxicity and ultimately resulting in neurodegeneration. Studies suggest that BSCL2 mutations can lead to a wide range of manifestations involving both upper and lower motor neurons, as well as peripheral motor axons. Patients with the P.N88S mutation may have a shorter disease course than patients with the P.S90L mutation. 81
The ERLIN1 gene codes for an ER protein known as ER lipid raft-associated protein 1. ERLIN1 forms a ring-shaped complex with ERLIN 2 to create the ERLIN1-2 complex. This complex is involved in the ER-associated degradation (ERAD) pathway, which is activated when the ER is under stress. The ERAD pathway identifies misfolded proteins, ubiquitinates them and transports them for degradation via a proteasome or autophagosome. Although limited to case series analyses, ERLIN1 mutations have been shown to correlate with SPG62, a pure subtype of HSP that specifically impairs upper motor neurons and causes focal deficits. 83 Given that ERLIN2 participates in the ERAD pathway of inositol 1,4,5-triphosphate (IP3), which is important for ubiquitination and the control of cellular cholesterol homeostasis, 84 further investigation into this connection is warranted, as the genetic spectrum of the disease may be broader than initially reported.
The SPG11 gene, responsible for the development of spastic paraplegia and the most prevalent subtype of HSP, is inherited in an autosomal recessive manner. It codes for a protein called Spastacsin, whose exact function is yet to be fully understood. 85 This genetic mutation typically results in progressive spasticity, neuropathy, cognitive impairment and a thin corpus callosum observed on MRI. The role of Spastacsin may also be involved in weight gain and management, but its complete range of functions remains unclear. 85 Exome sequencing has shown that heterozygous deletions may lead to missense and nonsense mutations, with premature stop codons in the SPG11 gene. Such mutations are prominently expressed across various brain regions, leading to nonsense, insertional and deletional mutations, resulting in frame shifts and loss of functionality. 86 This is particularly relevant because it has been associated with corpus callosum thinning, dementia symptoms and overall grey matter impoverishment. 85 Additionally, reduced volumetric size has been reported in various brain regions, such as the thalamus, accumbens nucleus, substantia nigra, amygdala and red nucleus. 85 Notably, clinical studies have shown that the spectrum attributed to SPG11 mutations is broader than previously documented, with patients afflicted by autosomal recessive juvenile ALS (ARJALS) exhibiting extended periods of survival. 87 Therefore, conducting a comprehensive analysis of SPG11 mutations in additional families with motor neuron disorders showing similar clinical manifestations is essential. 87 Consequently, further research into the mechanisms causing neurodegeneration associated with SPG11 functionality is warranted. 86
Monogenic PD/parkinsonism
Parkinsonism represents a group of neurological disorders characterised by symptoms such as rigidity, tremors and slowed movement. 88 A significant study of SPG11, one of the largest cohort studies on the subject, revealed that 60% of participants responded to levodopa (L-Dopa) treatment, and one in six SPG11 individuals exhibited parkinsonian features, suggesting a strong connection between parkinsonism and SPG11. 89 Research in this area has shed light on the crucial role of SPG11 in the existence and functionality of neurons. 89 Specific SPG11 mutations have been shown to harm dopaminergic neurons, suggesting that these mutations might lead to early-onset parkinsonism. 90 Furthermore, the analysis of HSP patients presenting with parkinsonism-like symptoms has revealed various additional variants in genes such as SPG7, FA2H and the ZFYVE26/SPG15 complex HSP genes. 90 Patients with SPG7 often exhibit symptoms of Parkinsonism. 91 Regardless of the number of mutant alleles, the type of variation, or the status of patient or carrier, pathogenic variants of SPG7 impact mtDNA homeostasis. Consequently, SPG7 plays a role in promoting mtDNA maintenance, and variations in this gene may result in parkinsonism due to anomalies in mtDNA. 91 Intriguingly, the study mentioned found that a genetic cause of parkinsonism was not identified in more than half of the included patients, hinting at the possible involvement of as-yet-unidentified genes in this condition. 90
The ATP13A2 gene encodes a P5-type transport ATPase protein found in lysosomes. 92 While its specific binding partners are not yet known, ATP13A2 expression has been associated with neuronal protection by preventing the accumulation of manganese.70,92 Loss of ATP13A2 function can lead to mitochondrial and lysosomal dysfunction in neuronal groups, contributing to the development of neurological diseases such as HSP and parkinsonism. 70 Whole-exome sequencing and homozygosity mapping have revealed a homozygous p.Thr512Ile (c.1535C 4T) mutation in ATP13A2, which has been associated with Kufor–Rakeb syndrome, an autosomal recessive form of juvenile-onset parkinsonism. This research underscores the connection between ATP13A2, parkinsonism and HSP. 70 Routine screening of patients with complex HSP for ATP13A2 mutations may prove beneficial in clinical practice.
The UCHL1 gene, also known as the PARK5 gene, codes for ubiquitin carboxyl-terminal esterase L1, which is a deubiquitinating enzyme involved in hydrolysing ubiquitin in the ubiquitin-proteasome pathway. UCHL1 is highly expressed in neuronal, neuroendocrine and podocyte cells, where it helps maintain ubiquitin stability, a crucial factor in brain functionality. 93 Meta-analyses of the UCHL1S18Y variant and its association with PD have demonstrated a significantly increased risk of sporadic PD in individuals with the AA genotype of this gene variant. 94
Hereditary neuropathy
Hereditary neuropathies encompass a class of inherited diseases affecting the peripheral nervous system. One such condition is CMT, characterised by symptoms such as symmetrical, length-dependent motor neuropathy, muscle weakness, paraesthesia and foot deformities. Typically, CMT manifests in childhood and progresses slowly. There are clinical and genetic similarities between CMT and other distal hereditary motor neuropathies (dHMNs). 92 Genes associated with dHMNs, including the COQ7 variant, have also been linked to HSP. 95
CMT type 2 is primarily caused by mutations in the BSCL2 gene. Exome sequencing studies have identified BSCL2 mutations in individuals clinically suspected of having CMT type 2. For example, among 47 Japanese CMT patients, 5 had heterozygous BSCL2 mutations. Notably, p.N88T and p.S141A mutations were observed in three cases. Patients with p.N88T exhibited a severe clinical presentation that began at an early age (1.5 years) and included vocal cord paralysis, distinguishing it from previous findings. 96 P.S141A is considered a unique and uncommon variation associated with CMT demyelinating neuropathy. 96 However, further cases are needed to confirm the pathogenic significance of p.S141A and p.N88T in CMT. In another study, three of 206 Chinese patients with CMT had BSCL2 mutations (p.S154W, p.S154L, p.A437P and p.A282V). Variants of the BSCL2 gene have also been implicated in dHMNs. In Taiwan, 76 CMT type 2 patients with dHMNs were found to carry two BSCL2 variants: p.S90L (in a CMT family) and p.R96H (in a sporadic dHMN case). 97
Methionyl-tRNA synthetase, also known as MARS1, is an enzyme responsible for charging tRNAs with their corresponding amino acids, a crucial step in protein synthesis.97,98 Mutations in MARS1 have been linked to the aberrant production of neurofilaments within axons. 99 Although MARS1 has been suggested as a potential gene associated with a subtype of CMT disease, the evidence is limited to exome sequencing analysis of a single family with CMT, necessitating further investigation into the potential role of MARS1 mutations, particularly in cases of axonal CMT.100,101
REEP1, encoded by the REEP1 gene, is a protein expressed in neurons throughout the brain and spinal cord, localised in both the ER and mitochondria. It is believed to play a role in developing the tubular network structure of the ER, regulating its size and protein processing capacity, and enhancing G-protein coupled receptor (GPCR) activity. 102 The HSP phenotype has been associated with REEP1. Interestingly, intellectual disability and behavioural problems have been linked to factors outside of REEP1 in HSP patients, such as SPAST-HSP missense mutations.102,103 Additionally, the SPG31 gene has been correlated with REEP1, with REEP1 recently identified as the causative gene for dHMN type 5. 104
The ALS5/SPG11/KIAA1840 gene has emerged as the causative factor behind a diverse array of clinical presentations, encompassing autosomal recessive HSP, ARJALS and autosomal recessive axonal CMT2 (ARCMT2). Notably, no significant large deletions or duplications have been associated with the ALS5/SPG11/KIAA1840 gene, and haematological and biochemical profiles appear unremarkable. 105 However, it is linked to the loss of myelinated fibres, resulting in motor and sensory axonal neuropathy, with a more pronounced impact on the lower limbs and characterised by low amplitudes of compound motor and sensory nerve action potentials. 105 As such, it is imperative to consider genetic screening of the ALS5/SPG11/KIAA1840 gene not only in patients exhibiting symptoms of autosomal recessive HSP with thin corpus callosum and ARJALS but also in cases of ARCMT2. 105
Given the intricate and heterogeneous nature of these neurodegenerative conditions, next-generation sequencing techniques stand as the most effective diagnostic tools for identifying the genetic underpinnings in patients presenting with myelopathy or neuropathy, whether through targeted sequencing panel approaches or whole-genome sequencing. 105
Cerebellar ataxia
Ataxia is a clinical manifestation characterised by issues such as speech difficulties, abnormal gait, loss of voluntary muscle control and uncoordinated ocular movements. Cerebellar ataxia patients often exhibit symptoms like tremors, overshooting movements, impaired articulation and poor balance. Importantly, these ataxia-related features share common pathogenic mechanisms with HSP. 106
CAPN1
The Calpain-1 (CAPN1) gene encodes calcium-activated neutral proteases known as calpains. CAPN1 is believed to contribute to essential neural processes and functional pathways that are common to both the corticospinal and cerebellar tracts. 107 Its involvement has been associated with SPG76, a condition characterised by cerebellar ataxia, further underscoring the connection between cerebellar ataxia and HSP.107,108
KIF1A
The KIF1A gene codes for a motor protein responsible for transporting membrane-bound organelles along axonal microtubules. Mutations in KIF1A have been linked to autosomal recessive SPG30 and, in some cases, are associated with cerebellar ataxia. Additionally, hereditary sensory neuropathy and mental retardation type 9 (autosomal dominant) have been observed in connection with KIF1A mutations.109,110 Notably, an estimated number of 61 KIF1A mutations have been reported, and certain missense variants have been associated with cerebellar atrophy and ataxia.110,111
Spastic ataxia of Charlevoix–Saguenay
Spastic Ataxia of Charlevoix–Saguenay (SACS), also known as Sacsin, encodes the sacsin protein highly expressed in the central nervous system. It plays a crucial role in recruiting Hsp70 chaperone proteins that help regulate the effects of ataxia-related proteins, such as amazon-1.112,113 Whole-exome sequencing studies have identified various SACS variants and their associations with changes in protein function and resultant phenotypes. For instance, gene variants p.Y508C and L.456V have been linked to infantile-onset spinocerebellar ataxia. 114
VPS13D
Vacuolar Protein Sorting 13 Homolog D (VPS13D) codes for a protein involved in lipid transport between organelle membranes and is thought to play a role in mitochondrial clearance, contributing to overall cell health, mitochondrial size and homeostasis. 115 Dysfunctions in the VPS13D gene have been reported in patients with autosomal recessive cerebellar ataxia. In some cases, individuals with VPS13D mutations have displayed adult-onset cerebellar ataxia, macrosaccadic intrusions, pyramidal tract signs and neuropathy. 116 In other instances, VPS13D mutations were found in individuals with pure HSP or complex HSP, with variations in the age of onset and symptom complexity associated with the nature of the mutations.117,118
ERAD pathway
The ER plays a pivotal role in the regulation of cellular calcium levels. Two distinct classes of calcium release channels, inositol IP3 and ryanodine receptors, are instrumental in mediating this regulation. GPCRs on the plasma membrane activate IP3 receptors located in the ER membrane, leading to the efflux of calcium from the ER into the cytoplasm. 119 Dysregulation of this mechanism has been implicated in the pathophysiology of several neurodegenerative diseases, including HSP, spinocerebellar ataxias and Huntington’s disease. The ERAD pathway is responsible for the degradation of active IP3 receptors. Compromised functioning of the ERAD pathway may also contribute to the development of neurodegenerative disorders. 119
The intersecting genes and metabolic pathways in diseases related to HSP have been summarised in Table 2.
Summary of intersecting genes and metabolic pathways in diseases related to hereditary spastic paraplegia.

HSP: hereditary spastic paraplegia; fALS: familial amyotrophic lateral sclerosis; SPG: spastic paraplegia; dHMN: distal hereditary motor neuropathy; DNA: deoxyribonucleic acid; CMT: Charcot–Marie–Tooth disease; ARJALS: autosomal recessive juvenile amyotrophic lateral sclerosis; ARCMT2: autosomal recessive axonal Charcot–Marie–Tooth disease type 2; ERAD: ER-associated degradation; IP3: 1,4,5-triphosphate.
Current and emerging therapeutic targets and management strategies
This review covers both pharmacological therapies and non-pharmacological therapies for HSP management.
Pharmacological therapies
Intrathecal baclofen therapy
Baclofen if a selective GABA-B receptor agonist and is frequently used for the treatment of spasticity. 120 It can be administered orally or intrathecally by the surgical implantation of a specialised pump. A more recent retrospective investigation of intrathecal baclofen (ITB) device implantation in seven patients with HSP revealed significant improvements in the Reflex Scale and spasticity according to the modified Ashworth Scale, as well as improvements in the modified Rankin Scale. 120 Long-term implantation observed improvement in spasticity for 2–3 years, followed by a stable phase of ambulatory and other mobility functions for 4–5 years. Additionally, there was no reported adverse events (Table 3). 120
Summary table of the effects of pharmacological treatments on hereditary spastic paraplegia.

HSP: hereditary spastic paraplegia; GABA: Gamma-aminobutyric acid; SPG: spastic paraplegia; MAS: MAS modified Ashworth scale; SPRS: spastic paraplegia rating scale, MFIS: modified fatigue impact scale, 6 MWT: 6-meter walk test; CYP7B1: oxysterol-7-hydroxylase; L-Dopa: Levodopa; MSWS-12: 12-item multiple sclerosis walking scale; TWT: time up and go test; 27-OHC: 27-hydroxycholesterol.
Levodopa
L-Dopa, an amino acid precursor to dopamine commonly used to manage PD symptoms, has been explored for its potential in HSP treatment. Historically, the evidence for L-Dopa in HSP has been largely based on case studies, offering limited-quality data.121,122 Although case studies have reported positive outcomes with L-Dopa treatment in HSP patients, an RCT conducted on individuals with SPG11 HSP-related parkinsonism did not show significant improvements in the cohort. 89 This disparity highlights the need for more comprehensive research to unravel the complex mechanisms underlying parkinsonism in HSP. For instance, the ineffectiveness of L-Dopa treatment might be attributed to extreme postsynaptic disruption, suggesting that further studies are warranted (Table 3).
Botulinum toxin injection
Botulinum toxin functions by inhibiting neuromuscular activity, binding to cholinergic nerve terminals, and reducing acetylcholine release. In a double-blind, randomised, placebo-controlled crossover trial (SPASTOX Trial), 55 HSP patients received either botulinum toxin type A (BoNT-A) injections or saline injections. 123 The results indicated that BoNT-A effectively reduced adductor tone, with transient and tolerable adverse events. However, it did not lead to significant functional improvements (Table 3). 123
Dalfampridine (4-aminopyridine)
Dalfampridine, an oral medication given at a dose of 10 mg twice daily for 15 days, yielded significant results in trials. It is a voltage-dependent potassium channel blocker designed to enhance walking ability in patients. 121 A study involving six patients showed a general improvement in motor function, as assessed through gait analysis, the time up and go test (TWT), the 6-meter walk test (6 MWT) and reduced fatigue based on the Modified Fatigue Impact Scale (MFIS). Patients also reported enhanced concentration during specific activities and perceived improvements in emotional stability. 121 Nevertheless, since these studies were uncontrolled, further controlled research is essential to provide stronger evidence regarding the safety and efficacy of dalfampridine for HSP patients (Table 3).
Cholesterol-lowering drugs
SPG5-HSP is caused by recessive mutations in the gene for oxysterol-7-hydroxylase (CYP7B1), resulting in the accumulation of neurotoxic oxysterols. 124 Cholesterol-lowering drugs, such as atorvastatin, have shown some potential in managing this subtype of HSP. A study involving 14 HSP patients revealed that treatment with atorvastatin led to a moderate reduction in plasma 27-hydroxycholesterol (27-OHC) levels, along with a significant decrease in total serum bile acids. This was associated with a relative decrease in ursodeoxycholic and lithocholic acids when compared to deoxycholic acid levels (Table 3). 124
Drugs inhibiting neddylation process
As previously elucidated, the diminished function of spastin has been associated with the pathogenesis of HSP. Investigations conducted in a spastin mouse model of HSP and utilising patient-derived cells have unveiled that the overexpression of Homeodomain Interacting Protein Kinase 2 (HIPK2) or inhibition of neddylation processes can effectively restores spastin levels and subsequently rescue neurite cells. 125 These insightful observations present the intriguing prospect of employing pharmacological interventions to inhibit neddylation-mediated degradation in neurons, thereby unearthing a novel and promising therapeutic target for addressing SPG4-HSP (Table 3). 125
Non-pharmacological therapies
Transcranial and direct current stimulation
Research into transcranial and direct current stimulation as potential treatments for HSP has been undertaken to alleviate symptoms and enhance patients' quality of life. Repetitive transcranial magnetic stimulation (rTMS) is a non-invasive technique that involves modulating neural networks by applying magnetic pulses. 126 Two RCTs have explored the use of rTMS. Although one trial, involving eight patients, showed improvements in lower limb spasticity, secondary outcomes such as lower limb motor function and short-term quality of life improvements did not significantly differ between treatment and control groups. 127 In the other trial, leg muscle strength improved, but spasticity in distal leg muscles did not significantly change. 126 Although there was speculation that rTMS could be paired with gait training to achieve greater functional improvements, an RCT on gait-adaptability training for HSP patients did not show significant benefits. 128 Notably, an RCT on transcutaneous spinal direct current stimulation (tsDCS), a non-invasive method for modulating spinal cord function, demonstrated reductions in spasticity and is proposed as a complementary strategy for patients not responding to alternative treatments (Table 4). 129
Summary table of the effects of non-pharmacological interventions on hereditary spastic paraplegia.
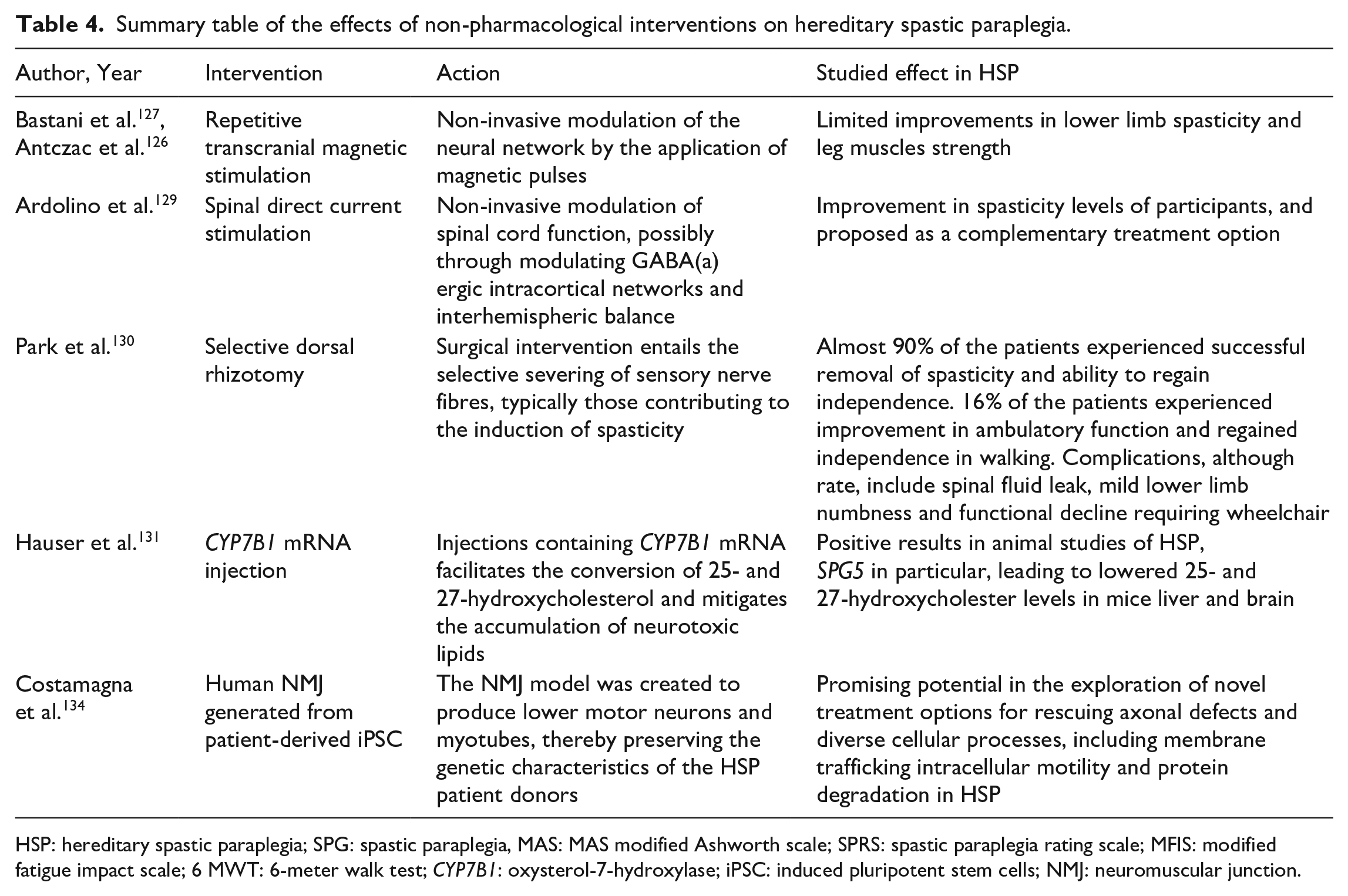
HSP: hereditary spastic paraplegia; SPG: spastic paraplegia, MAS: MAS modified Ashworth scale; SPRS: spastic paraplegia rating scale; MFIS: modified fatigue impact scale; 6 MWT: 6-meter walk test; CYP7B1: oxysterol-7-hydroxylase; iPSC: induced pluripotent stem cells; NMJ: neuromuscular junction.
Selective dorsal rhizotomy
Selective dorsal rhizotomy (SDR) is a surgical procedure commonly used to manage spastic cerebral palsy, aiming to reduce spasticity and enhance ambulatory function. It has also been explored for HSP patients. 130 In a study by Park et al., 130 37 HSP patients, with SPG4 and SPG3A being the most common genetic mutations, underwent SDR. The procedure successfully eliminated spasticity in almost 90% of the patients, enabling them to regain independence. Approximately 16% of the patients experienced an improvement in ambulatory function, regaining their ability to walk independently. Although complications are rare, they can include spinal fluid leaks, mild lower limb numbness, and functional decline necessitating the use of a wheelchair (Table 4). 130
Disease modifying therapies and biomarkers
Currently, there are no available disease modifying therapies (DMTs) for HSP. However, certain subtypes may present opportunities for the development of interventions. For example, SPG5, characterised by a loss-of-function mutation leading to the accumulation of 25- and 27-hydroxycholesterol, which can cross the blood–brain barrier and disrupt neurons, has been a focus of study. The injection of CYP7B1 mRNA has been explored as a potential treatment option. 131 This strategy has demonstrated a reduction in the accumulation of neurotoxic agents in mouse liver and brain. Given the potential for targeting this enzyme through lipid-lowering therapies, 132 it is conceivable that DMTs may be identified along this route in the future (Table 4). 131
Novel stem cells
The use of patient-derived induced pluripotent stem cells (iPSCs) has been investigated for various neurological and motor disorders, including spinal muscular dystrophy and ALS. However, HSP, characterised by more widespread axonal degeneration that extends beyond the corticospinal tract, may limit the potential application of iPSCs, as described in the literature. 133 Human neuromuscular junctions (NMJs) generated from iPSCs obtained from five HSP patients have shown promise. These NMJs create lower motor neurons and myotubes while maintaining the genetic background of HSP patient donors. 134 The NMJ model has displayed potential in exploring novel treatment options for addressing axonal defects and various cellular processes, including membrane trafficking, intracellular motility and protein degradation in HSP (Table 4). 134
Limitations of study
This comprehensive review employs a methodologically rigorous approach to elucidate various facets of HSP, offering valuable insights into its underlying mechanisms, genetic determinants and the current landscape of treatment options, including those under active investigation. Nevertheless, it is essential to acknowledge certain inherent limitations.
Firstly, the literature search strategy encompassed a diverse array of reputable databases, including Scopus, MEDLINE, EMBASE and the Cochrane Library. Despite the diligence in this process, it is possible that some pertinent studies on the subject may have inadvertently been omitted, as no search methodology can claim absolute exhaustiveness. Secondly, a notable limitation lies in the review’s exclusive focus on English-language literature. While this approach ensured the inclusion of a substantial body of relevant research, it may have unintentionally excluded valuable insights presented in non-English publications. This linguistic bias introduced by the language restriction warrants consideration, as it may have implications for the comprehensiveness of the review. Furthermore, the review process itself may introduce certain biases. For instance, the selection of specific keywords for the search query may inadvertently exclude relevant literature that employs differing terminology, thereby potentially affecting the scope of the review. Interpretive bias, stemming from the subjective judgement involved in synthesising and analysing the selected literature, is an additional limitation. Lastly, it is worth noting that some studies are small-scale with limited sample sizes, limiting the generalisability of the findings across the population.
Conclusion and prospects
Due to the heterogeneous clinical nature of HSP, early diagnoses and subsequent management becomes challenging. Although genetics-based management has shown progress for some neurological disorders, HSP lags behind due to its genetic diversity, complex disease processes, range of subtypes and slower disease onset. Moreover, screening and scoring systems, such as the 6- and 10-min walk test alongside the TWT used to assess fatigue and quality of life, may be beneficial in HSP. To advance our understanding and treatment of this neuromuscular condition, extensive research is required, which should involve international collaboration and RCTs conducted on substantial population cohorts. These efforts are particularly critical when considering the administration of combination medications. Furthermore, there is a pressing need for the identification of biomarkers and distinctive mechanisms that can differentiate HSP from other neurological disorders presenting similar clinical symptoms. Additionally, research should focus on the development of promising pharmacological and non-pharmacological treatments, striving to strike a balance between treatment efficacy and the mitigation of adverse effects when addressing HSP symptoms.
Footnotes
Acknowledgements
We acknowledge Icormed Research Collaborative and Toufik’s World Medical Association for their research facilitation.
Author contributions
Conceptualisation: W.A.A. and H.H. Data Curation: W.A.A., J.K.T., A.D.S., T.F., F.T.A., A.M., J.W., L.D., E.C. and H.H. Writing – Original draft: W.A.A., J.K.T., A.D.S., T.F., F.T.A., A.M., J.W., L.D., E.C., H.H., T.A.R., V.S., O.A., J.K., R.J. and D.E.M. Writing – Second draft: W.A.A., J.K.T., A.D.S., T.F., F.T.A., A.M., J.W., L.D., E.C., H.H., T.A.R., V.S., O.A., J.K., R.J., D.E.M., K.S., O.K. and M.Y.D. Review and Editing: W.A.A., J.K.T., A.D.S., H.H., O.A. and M.Y.D. Visualisation: V.S. and A.D.S. Approval of final manuscript: All authors.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable.
Informed consent
Not applicable.
Data availability statement
No data available.