
Abstract
Adult T-cell leukemia/lymphoma (ATLL) is a rare, human T-lymphotropic virus type 1 (HTLV-1)–driven neoplasm that is often underrecognized in low-endemic regions. We describe a 23-year-old man with an eight-year history of chronic dry cough who later developed weight loss, night sweats, odynophagia, and dyspnea. CT revealed cervical/supraclavicular lymphadenopathy with innumerable pulmonary micronodules, interlobular septal thickening, and ground-glass opacities; abdominal imaging showed hepatosplenomegaly, ascites, and extensive retroperitoneal/mesenteric adenopathy. HTLV-1 ELISA and confirmatory Western blot were positive. Excisional lymph node biopsy demonstrated diffuse architectural effacement by atypical T cells with a CD3+, CD4+, CD5+, CD25+, CD7–, granzyme B+ immunophenotype and a ~90% Ki-67 index, establishing lymphomatous-type ATLL with a cytotoxic profile. Despite rapid recognition, the patient deteriorated and died from respiratory failure 15 days after diagnosis, before chemotherapy could begin. This lung-predominant presentation in a young adult illustrates how ATLL can mimic chronic pulmonary disease and evade early detection outside endemic areas. Clinicians should prioritize early tissue acquisition and a minimal T-cell panel (CD3, CD4, CD25, CD7, Ki-67); a CD4+CD25+ phenotype with CD7 loss should prompt HTLV-1 testing irrespective of geography. Streamlined access to immunophenotyping and confirmatory HTLV-1 assays is essential to reduce diagnostic delays and improve outcomes.
Plain language summary
This report describes a 23-year-old man who had a dry cough for eight years. Over time he also developed weight loss, night sweats, trouble swallowing, and shortness of breath. Scans showed enlarged lymph nodes in the neck and abdomen and many small spots in the lungs. Common infections were ruled out. A surgical sample of a swollen lymph node confirmed a rare blood cancer called adult T-cell leukemia/lymphoma (ATLL). ATLL is caused by infection with a virus named human T-lymphotropic virus type 1 (HTLV-1). The virus is common in some parts of the world but uncommon in many others. Because of this, doctors outside high-risk regions may not think of ATLL right away—especially when symptoms seem to come from the lungs and blood tests are not striking. This can delay diagnosis. Our case shows that ATLL can look like a chronic lung disease for years, even in a young person. We highlight a practical approach that may speed up care in low- and middle-income settings: (1) consider ATLL when long-lasting cough is accompanied by weight loss, night sweats, or swollen lymph nodes; (2) obtain a tissue sample early; and (3) request HTLV-1 testing when suspicion is raised. Clear referral pathways to outside laboratories can help when advanced tests are not available locally. Despite recognition after the biopsy, our patient’s breathing worsened rapidly and he died before treatment could begin. Raising awareness of this unusual presentation may help clinicians reach the diagnosis sooner and improve patient outcomes.
Keywords
Introduction
Adult T-cell leukemia/lymphoma (ATLL) is a rare, aggressive malignancy of mature peripheral T lymphocytes caused by human T-lymphotropic virus type 1 (HTLV-1).1,2 Although HTLV-1 has a global distribution, endemic foci persist in southwestern Japan, the Caribbean, South America, and sub-Saharan Africa. Outside these areas, unfamiliarity with HTLV-1–related disease and low pretest suspicion frequently delay diagnosis.3,4 After a prolonged latency, ATLL manifests in four clinical subtypes—acute, lymphomatous, chronic, and smoldering—with the lymphomatous variant characterized by nodal infiltration and an unfavorable prognosis. Classically, a disease of older adults in endemic regions, ATLL may nevertheless arise in younger individuals and in settings perceived as low endemic, where the diagnostic pathway is less well trodden.3,4
Pulmonary involvement in ATLL is uncommon as the dominant initial presentation and is easily misattributed to infection or interstitial lung disease, particularly when hematologic abnormalities are subtle. 5 Imaging findings (e.g., centrilobular nodules, ground-glass opacities, or bronchovascular thickening) are nonspecific and overlap with common respiratory disorders, compounding delays. 5 Immunophenotypically, ATLL cells usually display a helper T-cell profile (CD3+/CD4+/CD5+/CD25+) with loss of pan–T-cell markers such as CD7, while cytotoxic marker expression is atypical.6,7 In non-endemic contexts, early tissue acquisition coupled with a minimal T-cell panel (including CD3, CD4, CD25, CD7, and Ki-67) and prompt HTLV-1 testing are therefore critical to shorten time-to-diagnosis.8,9 Beyond its characteristic immunophenotype, molecular studies have also reported increased RAB3GAP2 expression in ATLL compared with ALL, 10 supporting the contribution of host cellular dysregulation to disease pathogenesis.
We report a young adult with lung-predominant lymphomatous ATLL whose prolonged respiratory symptoms obscured recognition. The aim of this report is to highlight the diagnostic challenges of atypical, lung-first ATLL presentations in low-endemic settings, emphasizing the clinical trajectory, potential resource limitations affecting early immunophenotypic evaluation, and the risk of rapid fatal progression when diagnosis is delayed.
Case presentation
This case report is reported in accordance with the CARE guideline (Supplemental File 1).
A 23-year-old man from Cali, Colombia, with no significant past medical history, first developed a persistent, dry cough 8 years ago. Although occasionally accompanied by scant whitish sputum, his cough failed to respond to multiple courses of outpatient antibiotics. Repeated bronchoscopy and routine infectious studies remained negative, and he continued to harbor a chronic, unexplained pulmonary complaint.
Over the past 6 months, he began to experience systemic “B” symptoms—unintentional weight loss of approximately 10 kg and intermittent night sweats. Two weeks before presentation, he noted progressive odynophagia and intolerance to solid foods, accompanied by anorexia, dyspnea on moderate exertion, and occasional postprandial vomiting. These new symptoms severely limited his daily activities and prompted further evaluation.
On physical examination, he appeared cachectic and exhibited bilateral cervical lymphadenopathy, with submandibular and supraclavicular nodes of varying size. Auscultation revealed decreased breath sounds at the lung bases without crackles, and abdominal palpation demonstrated hepatosplenomegaly without additional masses. Initial laboratory studies showed leukocytosis (13,060 cells/mm3) with neutrophil predominance, microcytic anemia (hemoglobin 10.2 g/dL), hypoalbuminemia (3.3 g/dL), and markedly elevated lactate dehydrogenase (968 U/L), AST (83 U/L), and alkaline phosphatase (193 U/L). Serologic screening for HIV, hepatitis B and C, and syphilis returned nonreactive.
Given the persistent lymphadenopathy, systemic symptoms, and unremarkable infectious workup, serology for HTLV‑1/2 was obtained. HTLV-1/2 serology was performed using enzyme-linked immunosorbent assay (ELISA), followed by confirmatory Western blot according to manufacturer specifications. Both tests were positive for HTLV-1 antibodies. Polymerase chain reaction (PCR) was not available locally at the time of diagnosis, raising suspicion for ATLL. Computed tomography of the neck and chest revealed multiple enlarged lymph nodes in levels II–IV and the supraclavicular region (the largest measuring 16 mm), along with numerous randomly distributed pulmonary micronodules, interlobular septal thickening, and ground-glass opacities (Figure 1). Abdominal imaging confirmed hepatosplenomegaly, moderate free ascites, and extensive retroperitoneal and mesenteric lymphadenopathy. Esophagogastroduodenoscopy demonstrated low-grade esophageal candidiasis, erosive gastritis, and extrinsic compression of the gastric wall, findings attributable to progressive immunodeficiency and nodal expansion.
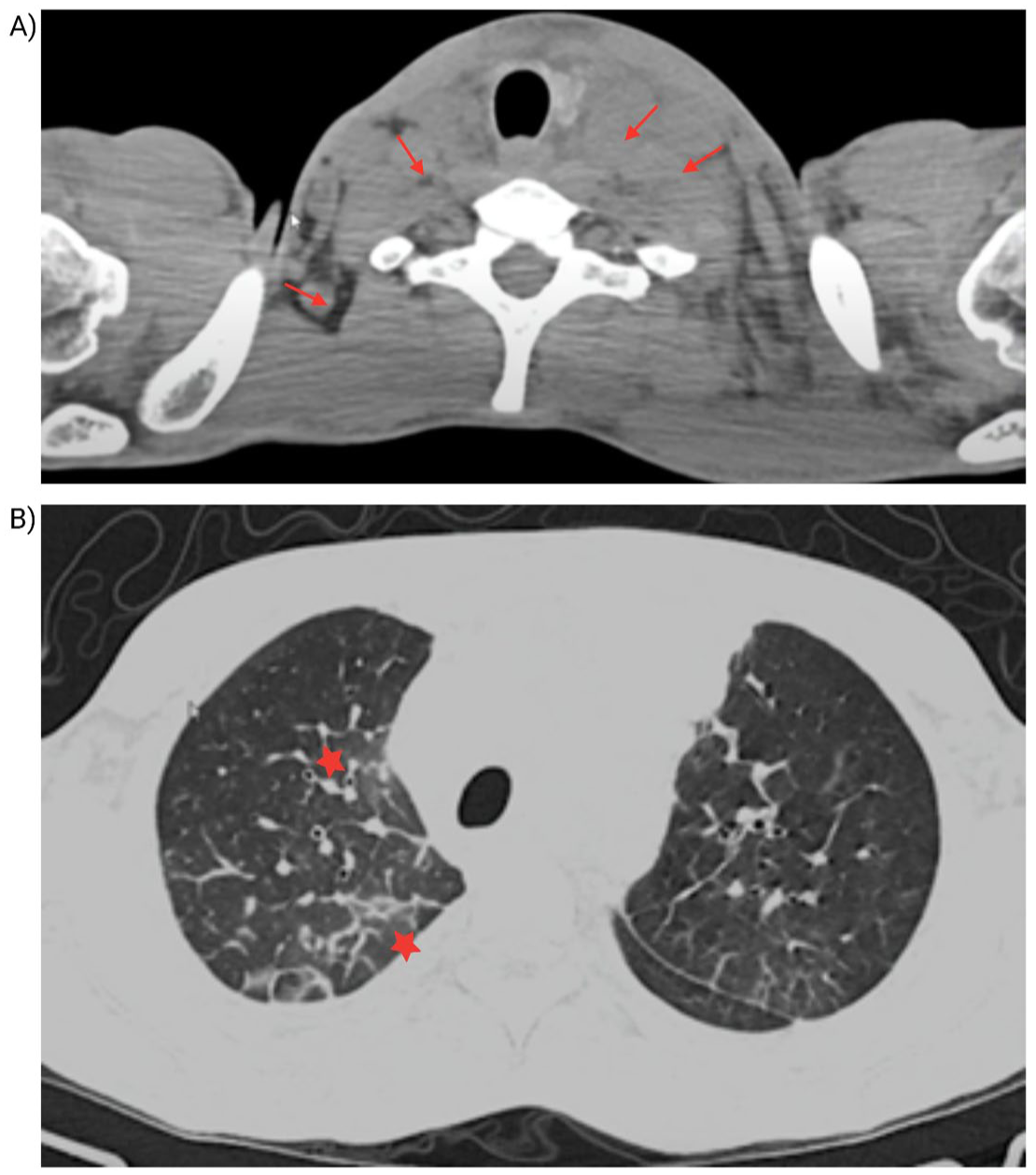
Axial computed tomography scans of the neck and chest. (a) Axial neck CT scan demonstrating multiple enlarged lymph nodes at levels II, III, and IVa (red arrows), including the supraclavicular region. The largest lymph node measures 16 mm on the left side. (b) Chest CT (lung window) showing randomly distributed micronodules, interlobular septal thickening, and both central and peripheral ground-glass opacities (red stars). Mediastinal lymphadenopathy is also observed, with a reactive appearance.
An excisional biopsy of a left supraclavicular lymph node showed diffuse effacement of the nodal architecture by lymphoid cells with irregular nuclei, prominent nucleoli, and numerous mitotic figures (Figure 2). Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue sections using standard automated platforms. Staining revealed strong, diffuse expression of CD3, CD5, and CD4, with loss of CD7 expression and high proliferative index (Ki-67). Markers for B‑cell lineage (CD20, PAX5) and other lymphoma-associated antigens (CD30, CD56, BCL2, BCL6, CD23, cyclin D1, MUM1) were uniformly negative, establishing a diagnosis of high‑grade peripheral T‑cell non‑Hodgkin lymphoma with a cytotoxic CD4+ phenotype consistent with the lymphomatous variant of ATLL.

Immunohistochemical analysis of cervical lymph node biopsy. (a) CD3 immunostaining showing diffuse and strong positivity (10×). (b) CD4 immunostaining with diffuse and strong expression (10×). (c) CD5 immunostaining also showing strong positivity (10×). (d) Granzyme expression demonstrating a heterogeneous pattern, indicating a cytotoxic T-cell component (10×). (e) Ki-67 staining revealing a high proliferative index (~90%), consistent with marked neoplastic activity (10×).
This case was managed in a regional public/social-security hospital where access to extended immunohistochemistry/flow cytometry and confirmatory HTLV-1 testing (Western blot/PCR) is intermittently available through external reference laboratories. Authorization, reagent shortages, and shipping logistics can delay comprehensive panels, which initially favored infectious differentials over early tissue diagnosis. Definitive classification required coordination with a reference lab and prioritization of a minimal T-cell panel, illustrating how resource realities shape diagnostic itineraries in the Global South.
Despite prompt recognition, the patient’s rapidly progressive respiratory compromise led to respiratory failure, and he died fifteen days after diagnosis—before chemotherapy could be initiated.
The chronological sequence of clinical events, diagnostic procedures, and disease progression is summarized in Figure 3.

Timeline of clinical course and disease progression in a patient with HTLV-1–associated lymphomatous-type ATLL. This timeline summarizes the patient’s clinical evolution, including the probable latent phase of HTLV-1 infection, onset of respiratory symptoms, emergency department and intensive care admissions, diagnostic procedures, confirmation of HTLV-1 infection and lymphomatous-type ATLL, rapid systemic deterioration, and fatal outcome.
Discussion
Adult T-cell leukemia/lymphoma is an uncommon, aggressive mature T-cell neoplasm caused by HTLV-1 infection. It is clinically divided into smoldering, chronic, primary cutaneous, lymphoma, and acute subtypes. The lymphomatous form typically presents with lymphadenopathy and little or no circulating leukemia; it is generally a disease of older adults in endemic regions (e.g., Japan, the Caribbean, and parts of Africa), and has a poor prognosis.11,12
Our patient’s age and isolated chronic pulmonary symptoms are highly atypical for ATLL. Nevertheless, ATLL cells most often express a helper T-cell phenotype (CD3+/CD4+/CD5+/CD25+) with loss of pan–T-cell markers such as CD7. The profound CD4+/CD25+ immunophenotype in our case is characteristic of ATLL, whereas the absence of CD7 further supports malignancy in an HTLV-1–associated T-cell process. Notably, the malignant cells expressed granzyme B, indicating a cytotoxic T-cell profile. This is unusual in ATLL: a recent immunophenotypic survey found that only about 2%–3% of cases express cytotoxic markers. Such cytotoxic marker–positive phenotypes may represent an aggressive variant of ATLL.1,11,12
The patient’s clinical course highlights the diagnostic challenge of ATLL presenting primarily with pulmonary symptoms. He reported a chronic dry cough for many years before systemic features emerged, without significant lymphadenopathy or leukocytosis initially. ATLL can cause respiratory symptoms: even indolent (smoldering or chronic) forms often produce pulmonary leukemic infiltration. 12 Yoshioka et al. 13 reported that ATL patients frequently complain of respiratory symptoms due to leukemic cell infiltration of the lung, even in smoldering/chronic phases. The clinical significance lies in recognizing that a persistent cough in young adults may be the sole sentinel sign of a highly aggressive and potentially fatal oncohematologic process (Table 1).
Lethal pathogenic sequence in lymphomatous-type ATLL associated with HTLV-1 infection.

Each stage represents a clinically and pathophysiologically interconnected progression, ultimately leading to multisystem deterioration and death in the absence of complete remission.
ATLL, adult T-cell leukemia/lymphoma; HTLV-1, human T-lymphotropic virus type 1.
However, isolated pulmonary involvement in ATLL is rare and easily mistaken for other conditions. Importantly, pulmonary abnormalities are highly prevalent in individuals with HTLV-1 infection regardless of ATLL status. In a large single-center cohort of 97 patients, Acuña-Villaorduña et al. 14 reported abnormal chest CT findings in 94.4% of patients with ATLL and 88% of those without ATLL. While lymphadenopathy and pulmonary nodules were more frequent in patients with ATLL, bronchiectasis and pleural thickening predominated in HTLV infection alone. These findings suggest that certain radiologic abnormalities—particularly ground-glass opacities, interlobular septal thickening, and bronchiectasis—may reflect HTLV-associated inflammatory lung disease rather than direct leukemic infiltration. In our patient, the extensive nodal disease combined with innumerable pulmonary micronodules favors lymphomatous involvement over isolated HTLV-related pulmonary inflammation, although both mechanisms may have coexisted.
Imaging in ATLL lung involvement can be non-specific: chest CT may show findings such as centrilobular nodules, bronchial wall thickening, ground-glass opacities or small consolidations, often accompanied by lymphadenopathy. For example, one series showed centrilobular nodules and patchy bronchovascular thickening in ATLL patients, changes that led to an initial misdiagnosis of infection (e.g., tuberculosis) in some cases. In our patient, persistent pulmonary infiltrates on imaging without a clear infectious etiology should have raised suspicion for an underlying malignancy such as ATLL, especially when accompanied by extrapulmonary findings (e.g., nodal enlargement or weight loss). The multiplicity of possible CT patterns and the overlap with infections often obscures the diagnosis. 5
Definitive diagnosis required a tissue biopsy. Bronchoscopic biopsies or needle biopsies can be inconclusive if only small samples are obtained. In fact, pulmonary lymphoma generally requires excisional or large core biopsy: prior reports note that small transbronchial samples frequently miss the diagnosis, necessitating surgical lung biopsy in many cases. In our patient, a comprehensive biopsy (likely from an involved lymph node or lung lesion) revealed the characteristic ATLL cell morphology and immunophenotype. Flow cytometry and immunohistochemistry were crucial: the aberrant CD4+/CD25+ T-cell population lacking CD7 strongly indicated ATLL, which was confirmed by HTLV-1 positivity. This aligns with established diagnostic recommendations that ATLL diagnosis relies on HTLV-1 serology (enzyme immunoassay and confirmatory Western blot or PCR) in conjunction with hematologic or histopathologic evidence of T-cell malignancy. Routine markers include CD3, CD4, CD7, CD8, and CD25, as used here. The unusual granzyme B positivity, as noted above, highlighted a cytotoxic variant; this immunophenotypic detail, though atypical, underscores the importance of broad panels in suspicious cases.11,15
Critically, HTLV-1 testing must be pursued even in non-endemic settings when clinical clues suggest ATLL. Our patient was likely living in or migrated to a region where HTLV-1 is not common. A recent series from Spain (a low-endemic country) reported that virtually all ATLL cases arose in immigrants or individuals with exposure to endemic areas. 16 Those authors recommend performing HTLV-1 serology at least once in all patients from endemic regions (or with partners from such areas) given the extremely poor prognosis of ATLL. Indeed, early recognition of HTLV-1 in atypical lymphomas can expedite diagnosis. In practice, when an ATLL-like immunophenotype is seen—especially CD4+CD25+CD7– T cells—clinicians should test for HTLV-1 antibodies, regardless of presumed geography. Delay in HTLV-1 testing can be fatal: symptomatic latency is long, and ATL may only become apparent decades after infection. 16
Beyond viral oncogenic mechanisms, host-related determinants likely contribute to ATLL progression. Differential expression of RAB3GAP2 has been described in ATLL when compared with ALL, 10 pointing to alterations in intracellular regulatory pathways. In parallel, immunogenetic evidence indicates that certain HLA polymorphisms may modulate susceptibility to malignant transformation among HTLV-1 carriers. 17 Collectively, these observations reinforce the concept that ATLL development reflects a multifactorial process rather than viral factors alone.
This case underscores that ATLL can masquerade as chronic lung disease. Non-leukemic pulmonary presentations (with or without subtle lymph node involvement) are documented but rare. HTLV-1–related pulmonary syndromes include lymphocytic alveolitis, bronchiolitis, bronchiectasis, and interstitial infiltrates. In established ATL, pulmonary infiltrates are usually attributed to opportunistic Ainfection or leukemic cell invasion. Our patient’s chronic dry cough likely reflected low-level pulmonary invasion by malignant T cells over many years. Only after an eventual systemic decline did the diagnosis of lymphomatous ATLL become evident. Similar diagnostic delays are reported for other “tissue-first” ATL cases (e.g., skin-first or lung-first presentations), where months to years elapse under non-malignant labels before biopsy confirms ATLL. 5
In practical terms, clinicians, especially in low- to mid-resource settings should retain a low threshold to consider ATLL when a young adult presents with chronic or recurrent respiratory symptoms accompanied by B-symptoms or otherwise unexplained lymphadenopathy, even in regions perceived as low endemic. Diagnostic momentum ought to favor early tissue acquisition; a minimal T-cell panel (CD3, CD4, CD25, CD7, Ki-67) can rapidly flag an ATLL-like profile, and the combination of a CD4+CD25+ phenotype with CD7 loss should trigger HTLV-1 testing irrespective of geography. Establishing streamlined referral pathways to reference laboratories for immunophenotyping and confirmatory HTLV-1 assays (serology and/or PCR) is therefore essential to prevent protracted, harmful delays in diagnosis and to align care with the operational realities of constrained health systems. Our case illustrates how adopting this sequence—early biopsy, targeted minimal panel, and prompt confirmatory testing—can shorten the time to a definitive diagnosis in presentations that otherwise masquerade as chronic lung disease.
A limitation of this report is the absence of an autopsy, which might have clarified the exact nature of pulmonary involvement. However, post-mortem examination is not routinely required for natural deaths in our setting. This highlights the importance of early ante-mortem tissue diagnosis in suspected ATLL.
Conclusion
The absence of classic hematological features such as florid lymphocytosis, combined with imaging findings mimicking chronic pulmonary or infectious diseases, led to a significant delay in diagnosis. Definitive identification relied on tissue biopsy and immunophenotyping that revealed a CD4+, CD25+, CD7– T-cell population with aberrant granzyme B expression—an unusual but increasingly recognized cytotoxic variant of ATLL. The positive HTLV-1 serology confirmed the retroviral origin of the malignancy. This case underscores the need to consider ATLL in the differential diagnosis of unexplained chronic pulmonary symptoms, even in non-endemic regions, particularly when associated with systemic signs and abnormal imaging findings. Early recognition and diagnostic suspicion are critical, as delays can result in rapid progression and fatal outcomes, as tragically occurred in this patient.
Supplemental Material
sj-docx-1-tai-10.1177_20499361261444136 – Supplemental material for Lymphomatous-type adult T-cell leukemia/lymphoma associated with HTLV-1 presenting as chronic respiratory illness in a young adult: a case report
Supplemental material, sj-docx-1-tai-10.1177_20499361261444136 for Lymphomatous-type adult T-cell leukemia/lymphoma associated with HTLV-1 presenting as chronic respiratory illness in a young adult: a case report by H. A. Nati-Castillo, M. Ocampo-Posada, Jhan S. Saavedra-Torres, Adriana Cecilia Correa Gomez, Marlon Arias-Intriago, Alice Gaibor-Pazmiño and Juan S. Izquierdo-Condoy in Therapeutic Advances in Infectious Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.