
Abstract
Background:
Gag protein of human immunodeficiency virus (HIV) has been reported to play a crucial role in establishing infection, viral replication, and disease progression; thus, gag might be related to treatment response. The objective of this study was to investigate molecular genotypes of the gag gene, particularly the important functional binding domains in relation to treatment outcomes.
Methods:
HIV-infected children enrolled and treated at Vietnam National Children’s Hospital were recruited in the study. A total of 25 gag sequences were generated and used to construct phylogenetic trees and aligned with a reference sequence comparing 17 functional domains.
Results:
We found that all patients in a treatment failure (TF) group belonged to one cluster of the phylogenetic tree. In addition, the rate of mutations was significantly higher in TF compared with a treatment success (TS) group, specifically the PIP2 recognition motif, and the nucleocapsid basic and zinc motif 2 domains [median and (interquartile range (IQR): 12.5 (6.25–12.5) versus 50 (25–50), p < 0.01; 0 (0–0) versus 0 (0–21.43), p = 0.03 and 0 (0–7.14) versus 7.14 (7.14–7.14), p = 0.04, respectively]. When analyzing gag sequences at different time points in seven patients, we did not observe a consistent mutation pattern related to treatment response.
Conclusion:
Gag mutations in certain domains might be associated with increased viral load; therefore, studying the molecular genotype of the gag gene might be beneficial in monitoring treatment response in HIV-infected children.
Introduction
Human immunodeficiency virus (HIV) disease progression following infection varies greatly between individuals. While certain patients remain asymptomatic for long periods of time without treatment, known as long-term non-progressors (LTNPs),1,2 the majority of patients follow a similar pattern of disease progression, with an increased viral load followed by viral suppression and the acquired immunodeficiency syndrome (AIDS) stage, during which the viral load is increased again. The duration of disease progression is dependent on a numbers of factors, in which HIV virus evolution and immune response are considered to be among the most important elements.3,4 Generally, children infected with HIV encounter faster rates of disease progression compared with that of their adult counterparts.5,6 Faster disease progression could be due to immature development of the immune system of children, and the substantial rate of evolution of the HIV virus in pediatric populations.3,5
Analyses of the HIV genome has revealed evidence of genomic instability, ranging from single-nucleotide polymorphisms (SNPs) to sequence deletions in HIV-1 genes encoding structural, regulatory, and accessory proteins such as gag (structural) and nef (accessory) in HIV-infected children.7,8 Diverse polymorphisms in the gag gene have been shown to be associated with disease progression, 9 whereas studies analyzing the HIV genome in long-term non-progressors LTNPs or “elite controllers” have reported no significant defects of the amino acid sequence of gag, 10 suggesting a role of genetic variability of gag in disease progression.
Gag protein, after being translated, is able to find and bind specifically to viral genomic RNA and bring this compound to the host cell membrane, facilitating viral budding from the membrane forming a new virion.11,12 Many studies have emphasized the irreplaceable role of gag protein in the process of viral assembly, binding, and maturation of new virions.13,14 Structurally, the gag polyprotein is stratified into four different big domains, including the N terminus matrix (MA), capsid (CA), nucleocapsid (NC), and the C terminus P6 domains, which are further divided into smaller domains with different functions in the HIV cycle process. The MA domain, which contains basic myristoylation, PIP (PCNA-interacting protein) 2 recognition motif, trimer interface 1, trimer interface 2, and nuclear localization 2 domains, is required for plasma membrane targeting, binding, and viral assembly. 15 The CA domain, which is responsible for development of a structural core, consists of NTD-NTD interface 2, NTD-NTD interface 3, cyclophilin A binding, MHR (major homology region), and dimerization domains. In addition, two important domains called nucleocapsid and zinc motif 2 belonging to the NC domain containing Zinc motif 1, have been shown to participate in recognition and interaction during viral replication. The last domain, P6, includes Vpr binding 1, ALIX interaction, and Vpr binding 2 domains and has been shown to be involved in processing viral particles.16,17 When these domains are mutated, viral replication, and, subsequently, disease progression might be significantly affected.18,19 With the current study, we investigated the molecular pattern of these functional domains in HIV-infected children.
Research methods and design
Setting
The study has a retrospective design. The study subjects were selected from outpatients diagnosed and treated at National Children’s Hospital, Hanoi, Vietnam. The treatment was first-line ART containing one non-nucleoside reverse transcriptase inhibitor (Nevirapine) and two nucleoside reverse transcriptase inhibitors (Stavudine or Zidovudine and Lamivudine).
Study population and sampling strategy
Patients were taken from a previous study 20 ; 86 patients were included, with full informed consent signed by the parents or responsible person. Ethical permission for the current research has also been approved by the ethical committee of Hanoi University of Public Health with the registration number 261/2015/YTCC-HD3.
In the study, 24 blood samples of patients were collected, and RNA was extracted and sequencing. The blood samples were used to determine CD4 T cell counts, CD4 T cell percentage, and HIV viral load. We later followed up patients but only managed to obtain seven samples after 24 months of following up for further analysis. The ID number of each patient, and status regarding treatment response, are included in supplemental data.
Laboratory methods
Peripheral CD4 T-cell counts were analyzed by Flow Cytometry (Sysmex Partec, Münster, Germany) and biannual quantification of viral load using the Cobas Taqman HIV-1 test (detection limit, 40 copies/ml).
Viral RNA was extracted from plasma samples (280 µl) by QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) and was used to synthesize cDNA using First-Strand Synthesis System for reverse-transcriptase (RT)–PCR (Invitrogen, Carlsbad, CA, USA) using random primers. A PCR was implemented to retrieve gag regions. The PCR mixtures and cycling conditions were described previously. 21 For the gag region, primers used to amplify HXB2 positions 796-2381 were F2NST (5′-GCGGAGGCTAGAAGGAGAGAGATGG -3′) and SP3AS (5′-CCTCCAATTCCCCCTATCATTTTTGG-3′). 22 First-round PCRs were conducted in 50 µl reaction containing 25 µl of master mix consisting of 10 × PCR Gold buffer (Applied Biosystems Inc., Foster City, CA, USA), a 40 µM concentration of each deoxynucleoside triphosphate (dNTP), 1.5 mM MgCl2, 0.75 U of AmpliTaq Gold DNA polymerase (Applied Biosystems Inc.); 2 µl of 0.4 µM concentration of each primer; and 5–10 µl of DNA template. The cycling conditions for the first round were 1 cycle at 95°C for 10 min; 30 cycles of 95°C for 10 s, the annealing temperature at 68°C for 30 s, and extension at 72°C for 1min; and a final extension at 72°C for 5 min. The second-round PCRs contained similar final concentrations in the PCR mixtures, but with 1 µl of the pooled first-round products with similar cycling conditions.
The final PCR products obtained were purified and directly sequenced using Big Dye terminator reaction kits and a capillary sequencer (Applied Biosystems 3100). The sequences obtained were aligned with reference sequence HXB2, with relevant subtypes and circulating recombinant forms (CRF01-AE), using Bioedit software and blast in NCBI and Mega 5.0 software. The gag gene sequence was further analyzed using the MAFFT version 7 program (https://mafft.cbrc.jp/alignment/server/). A total of 17 important functional domains were aligned with the HXB2 strain in the Los Alamos HIV database, including basic myristylation, PIP2 recognition motif, trimer interface 1, trimer interface 2, nuclear localization 2, NTD-NTD interface 1, NTD-NTD interface 2, NTD-NTD interface 3, cyclophilin A binding, MHR, dimerization, interaction domain, zinc motif 1, nucleocapsid basic domain, zinc motif 2, Vpr binding 1, and ALIX interaction. 8
Data analysis
Data were collected and managed by excel and analyzed by Stata 12.0.20 (Stata Corp LLC, College Station, TX, USA) for descriptive statistics and statistical inference. The descriptive statistic was used to summarize the variables of different groups, whereas the statistical inference was used to compare different variables between TF and TS groups. Since the data were not normally distributed, the descriptive statistics were median with interquartile range (IQR) and N (number) and % (percentage); whereas Mann–Whitney test and multiple comparison were used to compare different variables between treatment success (TS) and treatment failure (TF) groups.
Results
Characteristics of HIV-infected subjects
As can be seen from Table 1, the total participants were 25, in which 16 participants were male and 9 were female. The median age of HIV-infected children was 4.7 in the TS group (IQR 3.1–6.8), and 5.8 in the TF group (3.7–7.8). The median CD4 T cell count was not significantly different between TS and TF groups [594 (270–976) versus 523 (162–884), respectively]. Nevertheless, the median HIV viral load was significantly lower in the TS, compared with the TF group 400 (400–400) versus 5021,000 (42,000–10,000,000), respectively (p < 0.01). There were higher numbers of boys than girls in the TS group, whereas the TF group had opposite gender proportions; 80% of the patients in the TS group suffered from opportunistic infections, and all patients in the TF group had opportunistic infections, the majority of which were at clinical stage 2 and 3 (Table 1). There were six patients with a viral load of >1000 copies/ml and thus belonging to the TF group (the ID of patients include 1053, 1065, 1072, 1082, 1087, and 1145). The remaining patients belonged to the TS group (data not shown).
The general and clinical characteristics of HIV infected children in current study.

HIV, human immunodeficiency virus; IQR, Interquartile range; N, number; TF, treatment failure; TS, treatment success.
Genetic analysis of the gag gene in HIV-infected subjects
Phylogenetic analysis of the gag gene showed that all HIV from patients belonged to genotype CRF01_AE (data not shown). The result showed that all the patients in the TF group belonged to cluster 1 of the phylogenetic tree. Specifically, patients 1053, 1082, and 1145 belonged to cluster 1.1, whereas patients 1072, 1065, and 1087 belonged to cluster 1.2 (Figure 1). Three patients belonging to cluster 1.1 had high HIV viral load and low CD4 T cell counts, while patients belonging to cluster 1.2 had high CD4 T cell counts despite high HIV viral load.

Phylogenetic tree based on gag gene in HIV-1 infected participants in the study.
Stratifying into different functional domains of gag protein, we found that TF patients encountered a higher frequency of mutations compared with TS patients (supplemental Figure). While the majority of functional domains did not differ significantly between TS and TF patients, the median rate of mutations in the PIP2 recognition motif in the TF group was significantly higher compared with the TS group [50 (25–50) versus 12.5 (6.25–12.5), p < 0.01]. Similarly, nucleocapsid basic domain and zinc motif 2 also carried higher levels of mutations in TF compared with TS patients [0 (0–0) versus 0 (0–21.43), p = 0.03 and 0 (0–7.14) versus 7.14 (7.14–25), p = 0.04, respectively] (Table 2). When using multiple comparison, only PIP2 recognition motif and Nucleocapsid basic domain showed the significant different between two groups (p < 0.01 and p = 0.03, respectively).
The percentage of difference between different gag functional domain sequence of HIV-infected participant in the current study compared with the referenced sequence.
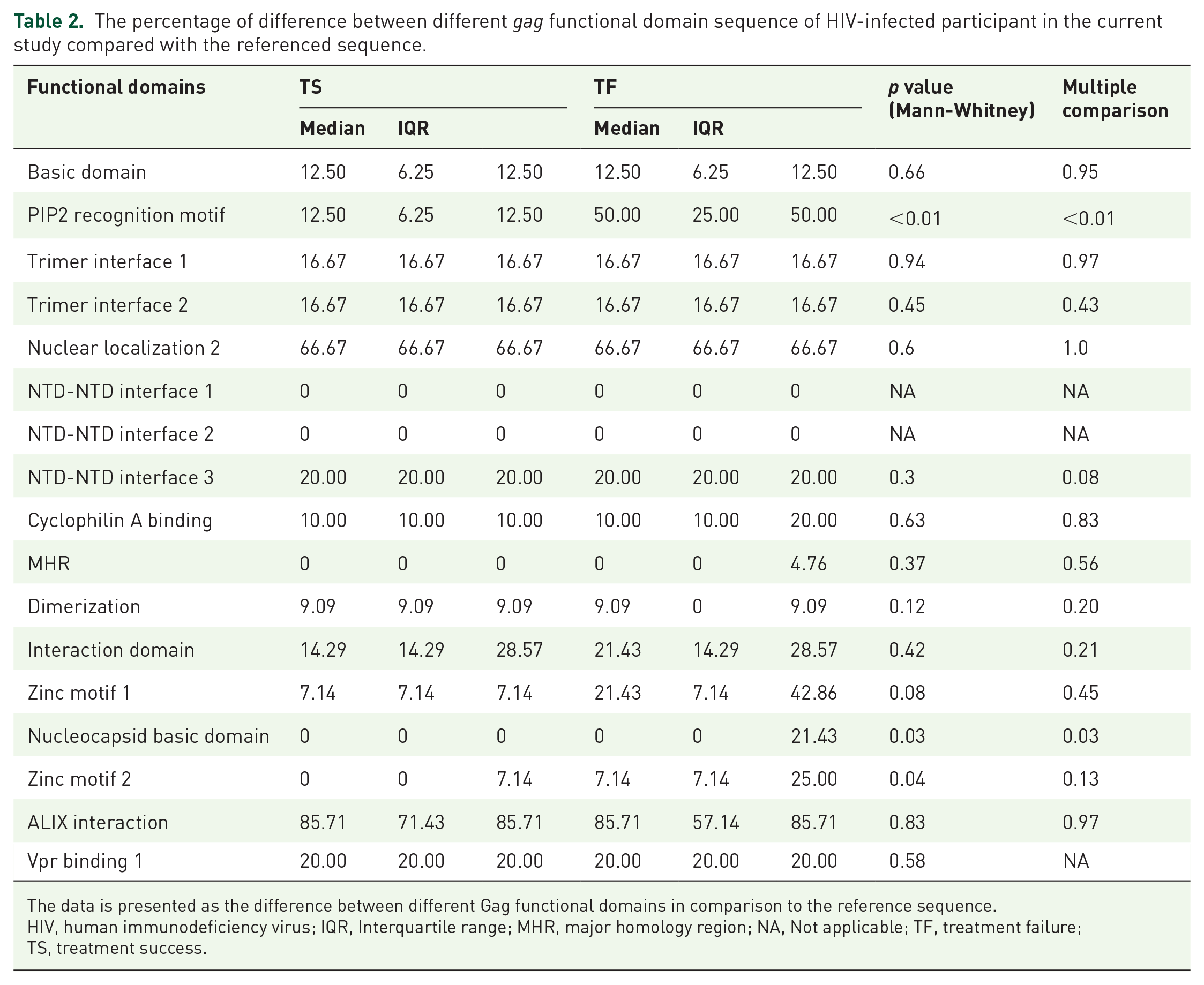
The data is presented as the difference between different Gag functional domains in comparison to the reference sequence.
HIV, human immunodeficiency virus; IQR, Interquartile range; MHR, major homology region; NA, Not applicable; TF, treatment failure; TS, treatment success.
Figure 2 represents the sequencing of seven patients with different time points. Regardless of the treatment response, all the sequences showed high levels of homogeneity compared with reference sequences. The trimer interface 2, and the NTD-NTD interface 3 domain sequences collected at different time points were highly homogenous, with only one amino acid substitution in both sequences, compared with the reference sequence. The results were similar for nuclear localization 2 domain, except for patients 1032, in which one amino acid was different between sequences taken at two different time points (Figure 2).

Mutations in the gag gene in HIV-1 infected participant in the study. The gag sequence of the same patients was analyzed at two different time points (L1 and L2, in which L1 was taken prior to treatment initiation while L2 was taken after 24 h of the treatment initiation). The two sequences at different time points were aligned with each other and aligned with the referenced sequence.
The cyclophilin A binding domain of patient 1058 showed one amino acid difference in both sequences compared with the reference gene, while patient 1082 was found to have one amino acid substitution within sequences taken at different time points. The zinc motif one domain showed one amino acid substitution in patients 1015; six amino acid substitutions in patients 1077, and one amino acid substitution in patient 1087, compared with sequences of the same patient taken at earlier time points. Both patients 1015 and 1087 had one amino acid insertion and one amino acid substitution in both sequences in comparison with the reference gene (Figure 2).
Discussion
In this study, we analyzed sequences of the gag gene in HIV-infected children in relation to treatment response. Consistent with most of findings concerning the HIV genome in South East Asia, our results confirmed presence of the subtype CRF01_AE in all patients in the present study. 23 Extensive studies have been conducted to investigate the association between HIV subtype and disease progression; however, the results have been inconsistent. 24 Saina et al. indicated that patients could be either rapid or slow progressors regardless of subtype distribution. 25
In the present study, we found that, even though all patients were found to carry the subtype CRF01_AE, phylogenic cluster analysis reveal that participants belong to different clusters of the phylogenic tree. In addition, Louwagie has suggested that the subtype A has the most diversity of gag gene among other subtypes 26 ; thus, we might speculate that the subtype CRF01_AE might encounter high levels of diversity. Since all children in the study were infected with HIV vertically, the HIV genotype reflected directly the genotype of their mother, suggesting the variety of complex recombinant forms of HIV in Vietnam. Interestingly, TF patients with high CD4 T cell counts and high viral load belonged to one cluster, whereas TF patients with low CD4 T cell counts and high HIV viral load belonged to another cluster. High viral load might be the result of the increase of viral replication, infectivity, viral fitness or/and immune-escape mutations. The results might implicate that HIV virus in these patients underwent similar mutations in the gag gene, leading to increased viral replication. Two patients (1053 and 1087) on the same cluster were shown to have developed reverse transcriptase inhibitor (RTI)-resistance mutations, which are the most dominant lamivudine-resistant mutations found among HIV infected children in Vietnam. 20 The M184V/I mutation has been found to be associated with increased viral load in several studies,27,28 and immunological recovery despite virological failure has previously been reported in patients with an M184V escape mutation that confers lamivudine resistance. 29 The mutation M184V/I has been shown to affect other mutations in different genes, 30 and it might be speculated that M184V/I might be associated with certain mutations in the gag gene facilitating increased viral load. Similarly, other mutations in the gag gene might result in lower viral load. The causal relationship between gag mutations and CD4 T cell counts has not been fully established. Therefore, the low CD4 T cell counts in the TF group belonging to a lower cluster of the tree might not reflect a direct consequence of profound viral replication as the result of gag mutations. Nevertheless, these patients might already suffer from low CD4 T cell counts prior to elevation of viral load as the result of gag mutations. Other clusters containing patients with high CD4 T cell counts despite high levels of viral load might be an interesting group to study. However, we have not found consistent mutations in this group, suggesting complexity of mutations and interaction between these mutations for the establishment of viral replication in different settings with CD4 T cell counts.
Saina et al. found no significant between-group differences in the amino acid variations, insertions, or deletions of gag sequences, and proposed that gag sequence variations are not as important as HLA in influencing disease progression. 25 Inconsistent with this finding, we did observe a significant difference in rate of mutations in the gag gene of the TF group in comparison with those of the TS group, particularly with regard to the PIP2 recognition motif and nucleocapsid basic domains. The PIP2 recognition motif is involved in the assemblance of HIV at the plasma membrane of infected cells, and several studies emphasize the importance of the PIP2 recognition motif for the production of infectious virions, 31 and, without the PIP2 recognition motif, membrane binding would not occur.32,33 Unfortunately, we could not find the established association between mutation of the PIP2 recognition motif and increased viral replication. Nevertheless, given the role of this domain, it is reasonable that high levels of mutations might be associated with increased HIV replication or infectivity in patients. Monde et al. pointed out that the PIP2 domain is crucial for Gag binding to the plasma membrane and the release of virus from the cell line. The adapted mutant virus (74LR) displayed accelerated replication kinetics compared with wild-type virus, which is probably due to increased virus infectivity. 34 We did not observe the 74LR mutation in our patient, suggesting that another mechanism might be involved in our cohort.
The nucleocapsid domain, on the other hand, is required for RNA binding activity and thus affects virus assembly.35,36 Several studies have pointed out the crucial role of the nucleocapsid domain for RNA packaging and recombination, and show that mutations at the nucleocapsid domain lead to inefficient package of viral RNA. However, in our data, mutations at this domain were observed in patients with high viral load, suggesting that these mutations might be associated with viral packaging and thus viral assembly. Regarding the mutations observed in the zinc finger motif domain, and consistent with our finding, Mark-Danieli et al. also identified a relationship between zinc finger mutants and increased RNA-binding specificities, and showed that the N17K mutant led to a 7- to 9-fold increase in RNA packaging. 37 Compared with the N17K mutant virus, E21K and N27K mutant viruses were not significantly superior to the WT virus in transduction efficiency. The domain has proven necessary for different steps of the HIV life cycle, and deletions of the domain might result in defective viral formation. The different mutations in the domain were found to yield different outcomes regarding viral development.37,38 We found that HIV mutations in these domains might be associated with increased HIV replication, and, thus, might enhance infectivity. Through evolution, certain mutants became associated with increased infectivity and others became associated with decreased viral stability. Understanding the interaction between these mutations is a complex issue in need of further research.
Overall, our results also confirmed that TF patients had higher levels of mutation in the gag gene in comparison to TS patients, which is consistent with the finding that patients with disease progression tend to have diverse polymorphisms in the gag gene, 9 while LTNPs or “elite controllers” showed limited mutations in the gag gene. 10 In addition, TF patients in the study also showed high levels of viral load, suggesting that gag mutations and viral load might have a certain association. The association between these two factors should be studied further in order to answer the question of how and why increased gag mutations might lead to increased pace of disease progression.
Our study has several limitations. First, we accessed mutations of gag sequences at only one time point and the number of patients was limited. Secondly, we were able to access only seven patients at different time points. However, given the diversity in the gag gene, our cases contribute to the understanding of mutations within the gag gene in relation to treatment response. Thirdly, in our setting, we could assess only the most prominent clone of HIV virus as this might represent the minority effect of HIV to disease progression, thus careful interpretation of the data should be taken into consideration. And finally, our results present observation only, therefore it is difficult to draw any evidence-based conclusions from the results.
Conclusion
In conclusion, Gag mutations in certain domains might be associated with increased viral load; therefore studying the molecular genotype of the gag gene might be beneficial in monitoring treatment response in HIV-infected children.
Supplemental Material
Supplementary_data_1 – Supplemental material for Molecular genotypes of gag sequences in HIV-1 infected children treated with antiretroviral therapy in Vietnam
Supplemental material, Supplementary_data_1 for Molecular genotypes of gag sequences in HIV-1 infected children treated with antiretroviral therapy in Vietnam by Linh Vu Phuong Dang, Hung Viet Pham, Thanh Thi Dinh, Phuong Thi Vu, Lam Van Nguyen, Hai Thanh Le, Mattias Larsson and Linus Olson in Therapeutic Advances in Infectious Disease
Footnotes
Acknowledgements
We would like to express our appreciation to the National Foundation for Science and Technology Development, Ministry of Science and Technology (grant number 106-YS.02-2014.22.), for financial support. We thank our students Nguyen Manh Tien, Nguyen Huu The Tung, Tran Thi Anh, Nguyen Anh Dung for valuable contributions. We would also like to thank The Swedish Foundation for International Cooperation in Research and Higher Education, STINT (through the TRAC Collaboration Sweden-Vietnam) for the support.
Author contributions
Linh Vu Phuong Dang: Conception and designing study; writing manuscript
Hung Viet Pham: performing experiment
Thanh Thi Dinh: literature search; administrative, technical and logistic support, performing analysis
Phuong Thi Vu: analysis and interpretation of the data
Lam Van Nguyen: sample and data collection
Hai Thanh Le: Provision of data and sample of patients
Linus Olson: Writing manuscript, critical revision of the manuscript
Conflict of interest statement
All authors were involved in the research and the writing of the manuscript and have approved the final version for this publication. We declare no conflict of interests.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Nafosted, National Foundation for Science and Technology Development, Training and Research Academic Collaboration (TRAC) – Sweden – Vietnam
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.