
Abstract
Summary points
• Individual variability in pain perception and differences in the efficacy of analgesic drugs are complex phenomena and are partly genetically predetermined.
• Analgesics act in various ways on the peripheral and central pain pathways and are regarded as one of the most valuable but equally dangerous groups of medications.
• While pharmacokinetic properties of drugs, metabolism in particular, have been scrutinised by genotype–phenotype correlation studies, the clinical significance of inherited variants in genes governing pharmacodynamics of analgesics remains largely unexplored (apart from the µ-opioid receptor).
• Lack of replication of the findings from one study to another makes meaningful personalised analgesic regime still a distant future.
• This narrative review will focus on findings related to pharmacogenetics of commonly used analgesic medications and highlight authors’ views on future clinical implications of pharmacogenetics in the context of pharmacological treatment of chronic pain.
Keywords
Introduction
The aim of any medical treatment is to individualise therapy, boosting the efficacy and minimising potential toxicity. It is estimated that response to over 25% of common medications, including analgesics, is influenced by some type of genetic variation, knowledge of which could be useful to prescribers. Furthermore, variation in drug efficacy may vary from 2- to 10-fold or even 100-fold among members of the same family. 1 –3 A similar pattern emerges when considering the frequency and intensity of side effects. 4
The completion of the Human Genome and HapMap projects, together with advances in high-throughput genotyping, has revolutionised our understanding of the importance of genetic predisposition and environmental variables, such as diet and general state of health, in individual drug responses.
According to the recent National Centre for Health Statistics report, analgesics constitute the most dangerous group of medications used legally. Opioids alone attribute to more than 15,000 fatalities annually with 343,000 emergency medicine (EM) department attendances in the United States alone due to drug overdose.
Pharmacogenetics is commonly defined as the study of genetic variation that gives rise to differing responses to drugs. More recently, another term has been introduced, pharmacogenomics, which covers the broader application of genomic technologies to new drug discoveries and the further characterisation of older drugs. Some authors use those two terms interchangeably; therefore, a clear distinction, as well as consensus definitions, needs to be agreed upon. 5 Behind the emergence of pharmacogenetics as a separate specialty lies our desire to understand how heritability affects the difference in responsiveness of different people to therapeutic agents.
Molecular genetics
The amino acid sequence of every protein is encoded by nucleotides, which form deoxyribonucleic acid (DNA). 6 Clearly identified DNA regions function as templates for the synthesis of messenger RNA (mRNA), and this process is called transcription. Messenger RNA leaves nucleus and is transported to the ribosome. Here, the RNA sequence is translated into a specific amino acid. Later, during the process of folding, the polypeptide chain of amino acids is transformed into an active molecule and then trafficked towards its final destination. A detailed description of the genetic code and the central dogma of molecular biology 7 is beyond the scope of this review but can be easily found elsewhere. 8 –10
Genetic polymorphism
A genetic polymorphism occurs when two or more distinct genotypes exist in the same population of a species. Polymorphism is also a main driver of natural selection and evolution. A single-nucleotide polymorphism (SNP) is a variation in the sequence of DNA when a single nucleotide (adenine (A), guanine (G), thymine (T) or cytosine (C)) differs between members of the same species. In human population genetics, it has been noted that the prevalence of certain SNPs can differ substantially between different ethnicities. SNPs can be inherited or occur de novo. Within a gene, an SNP may occur in intronic, non-coding regions or exonic, coding regions, where the change may be synonymous (no difference in amino acid sequence) or non-synonymous (alters the amino acid sequence). A non-synonymous SNP may lead to protein truncation (nonsense mutation) or affect folding or biophysical properties (missense mutations) and therefore may have important functional consequences. The Online Mendelian Inheritance in Man (OMIM) database defines the relationship between polymorphisms and diseases. 11 The Single Nucleotide Polymorphism Database (dbSNP) gathers information about minor genetic variation in the growing list of species. 12 In 2012, dbSNP contained over 187,000,000 SNPs in humans.
SNP nomenclature
At least three different ways of identifying genetic polymorphisms exist. All changes can be reported at the coding DNA-level (cDNA). For example, the SCN9A abbreviation c.2572C>T identifies a C to T substitution at position 2572 of the SCN9A gene in exon 15. 13 This is an example of the missense mutation, which leads to the replacement of leucine with phenylalanine in the alpha-subunit of the Nav1.7 sodium channel at amino acid position 858 (p. 858 Leu > Phe). The same SNP can be referred to by its dbSNP allocation number (rs80356476). In addition, nomenclature of all of the wild-type P450 enzymes is different. By convention, CYP2C9, for example, identifies three different variants labelled accordingly as CYP2C9*1, CYP2C9*2 and CYP2C9*3. Subjects may be homozygous or heterozygous for a particular allele; their genotype can be recorded as CYP2C9*3/*3 (homozygous) or CYP2C9*1/*3 (heterozygous).
Copy-number variations
Copy-number variation (CNV) is an example of a more significant alteration in the genetic code. Deletions, insertions and inversions are some other possible alterations that can occur. CNVs may involve an abnormal number of copies of the same gene or a complete deletion of the region. This is very common and up to 0.4% of the genome of two individuals differs with respect to CNVs. 14 The P450 2D6 enzyme gene CYP2D6 CNV, for example, can produce a complete CYP2D6 gene deletion (CYP2D6*5) or duplication (CYP2D6*x2), which can result in the reduced or increased metabolism of many xenobiotics. 15
Heritability of pain traits
Heritability estimates derived from inbred strains of laboratory animals suggest that up to 30–76% of the variance in pain behaviour can be explained by genetic influences. 16,17 A variety of chronic pain conditions, including sciatica, irritable bowel syndrome and mechanical back pain have been studied utilising human twin-studies. Some important initial estimates of trait variants attributed to the inherited genes were made based on this work. 18 –20 Individual variability attributed to the genetic factors in twins was further studied with experimental pain induced by a variety of noxious thermal and chemical stimuli in twins. Up to 50% of variability in experimental pain sensitivity was attributed to inherited factors. 21 In normal individuals, as well as chronic pain sufferers, it is not easy to correlate one cohort of twin subjects to the other, as it appears that the influence of genetic variables cannot be generalised from one pain state to another. The same generalisation can equally apply to data extracted from the experimental pain modalities.
The development of chronic pain is a good example of gene and environment interaction. Only a small minority of all individuals exposed to a noxious event, such as inflammatory or traumatic nerve tissue injury, actually develop chronic pain. 22,23 Once the acute or chronic pain has occurred, pain severities, 24,25 as well as responses to analgesics, can also be very variable among sufferers. 26,27 Both experimental and clinical twin and family studies in humans have contributed to our understanding of altered pain behaviour and variability in the intensity of response to the same stimulus. 28,29
An overview of peripheral pain circuitry and genes responsible for the variety of anatomical entities has been well described. 30
Pain genes
Association and linkage human studies have identified a number of genes responsible for heritable conditions involving dramatic alterations in pain perception. The hereditary sensory and autonomic neuropathies I–IV (HSANs I–V) are examples of a family of syndromes in which pain perception and responses are reduced or absent as the result of single point mutations. 31 More recent scientific discoveries have confirmed a pivotal role for the sodium channel Nav1.7 in a growing range of human familial and de novo gain-of-function and loss-of-function pain syndromes. 32,33 Gain-of-function lesions in SCN9A were identified in three distinct disorders: primary erythromelalgia (PEM), paroxysmal extreme pain disorder (PEPD) and idiopathic small fibre neuropathy (SFN). 33 –35 To date, 14 PEM mutations have been identified, most of which map to the first three domains of Nav1.7 36 Human monogenic pain syndromes provide important insights into the molecular mechanisms that underlie normal and pathological pain states. 37 Gene mapping of human mutants carrying an extremely altered pain phenotype spectrum, ranging from a complete loss of pain 32,38 as well as severe gain-of-function variants, such as inherited erythromelalgia, 33 has provided an exceptional opportunity to study key molecular mechanisms that are involved in the regulation of pain signalling. The knowledge thus obtained may be used towards a better understanding of the wider patient population affected by numerous chronic pain conditions.
Pharmacological concepts applied on pharmacogenetics
Figure 1 schematically portrays the function of different genes in influencing pharmacokinetic and pharmacodynamic properties.
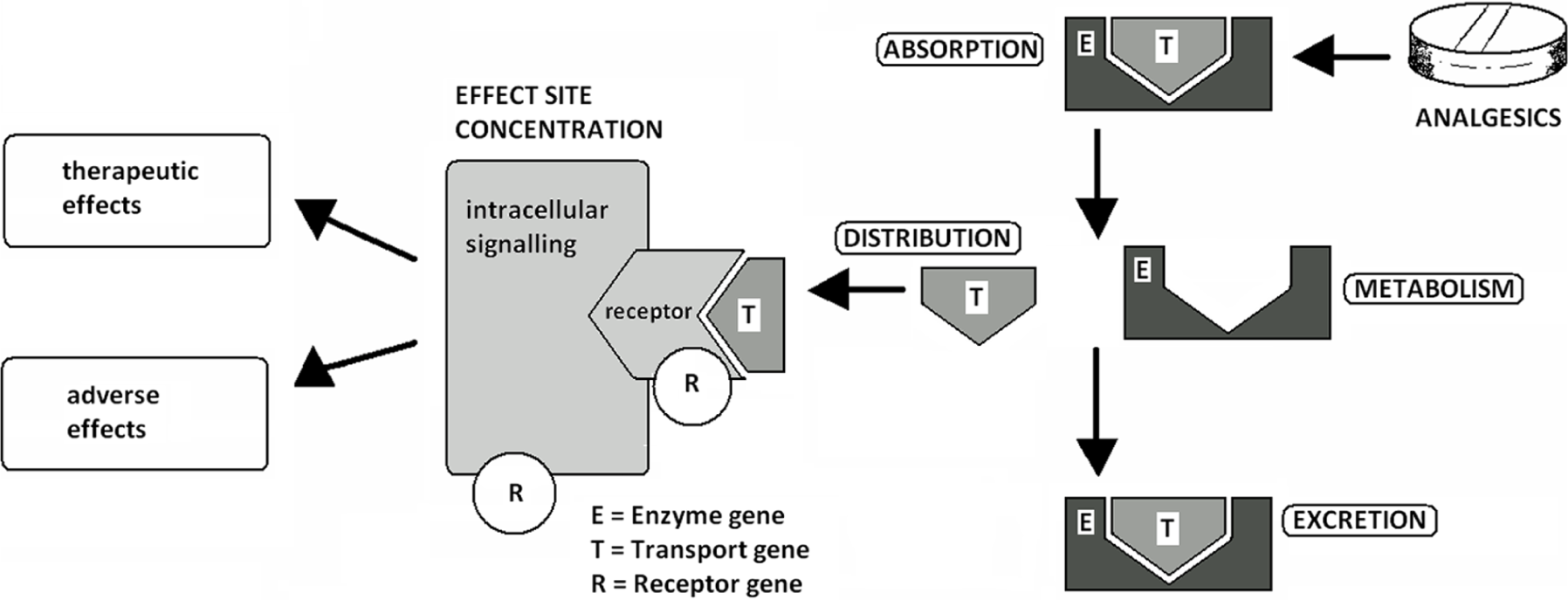
Heritable factors influencing drug–organism interaction.
Pharmacodynamics is the study of the effects of a drug on the human body. When describing different analgesic classes, the pharmacodynamics section of this article will focus on the polymorphisms in amino acid sequence variations of cell-surface proteins such as receptor and ion channels as well as SNPs in genes coding for various intracellular signalling pathway structures. Pharmacokinetics is supplementary to pharmacodynamics and is the study of drug absorption, distribution, metabolism and drug excretion. The aim of metabolism is to make molecules more water soluble and ready for renal or other excretion. Corresponding sections will look into how gene variations affect the metabolism of various pro-drugs as well as active metabolites. It will also investigate what effect genes have on the development of side effects of commonly used analgesics. We will start our review with the strongest and arguably most valuable group of analgesic drugs available – opioids.
Pharmacogenetics of opioid analgesics
Opioids are routinely used in the treatment of moderate or severe acute and chronic pain. There are several alternative strong opioids available, for example, morphine, oxycodone, hydromorphone and fentanyl, each with comparable efficacy at a population level. 40 At an individual level, however, there is wide variation in opioid analgesic efficacy and side effects, the reasons for which are not fully understood, but may in part be genetic. In cancer-related pain, up to 30% of patients do not respond well to morphine, either due to inadequate pain relief and/or intolerable side effects. The majority of these morphine ‘non-responders’ achieve a better clinical outcome when given an alternative opioid. 41,42 Common adverse events evoked by opioids include nausea and vomiting, drowsiness, confusion and hallucinations.
Pharmacodynamics
Opioid receptors
Opioid receptors belong to the family of G-protein-coupled receptors (GPCRs). There are three types of classical opioid receptor: mu (µ), kappa (κ) and delta (δ). Structurally similar, they contain an extracellular N-terminus, seven transmembrane domains and an intracellular C-terminus, and each share a high degree of homology. Most variation is found in the N-terminal domain and extracellular loops. 43,44 The extracellular loops determine ligand binding and are therefore particularly important. Splice variation of opioid receptor mRNA has been shown to produce receptor subtypes, which may be of functional importance. 45
Gene knockout studies in mice have demonstrated that analgesic response to opioids is primarily mediated by the µ-opioid receptor. 46 Genetic variation in the human µ-opioid receptor gene (OPRM1) has been associated with opioid response in several different clinical settings, including acute post-operative pain, 47 –49 chronic non-cancer pain 50,51 and cancer-related pain. 2,52
The non-synonymous exonic SNP c.118A>G (rs1799971) is the most consistently reported example of association between genetic variation in OPRM1 and opioid response. This SNP results in an asparagine-to-aspartic acid change at position 40, a putative N-glycosylation site in the important extracellular N-terminal region; however, the functional significance remains uncertain. 50 The variant G allele of c.118A>G has been associated with increased dose requirements of morphine in cancer patients 2,52 and patients following surgery. 47 –49 Similarly, the common A allele has been associated with improved analgesia from morphine in cancer-related pain. 52 Nevertheless, when these opioid pain studies were combined in meta-analysis, no association with increased pain was found, and only a weak association with increased morphine dose requirements in homozygous carriers of the variant G allele. 53 c.118A>G has also been associated with the opioid-related side effects. In one post-operative study of patients receiving morphine, carriers of the variant G allele had less sedation and less nausea. 54 Another post-operative study of intrathecal morphine and one of tramadol for osteoarthritis also reported an association with the variant G allele with less nausea/vomiting. 49,54,55 The c.118A>G genotype was, however, not associated with fentanyl-induced post-operative nausea and vomiting in another study of post-operative pain. 56 The pattern of less analgesia, in addition to less side effects (upper gastrointestinal and central), suggests reduced receptor sensitivity to opioids.
Other SNPs from OPRM1 and the other classical opioid receptor genes, including OPRK1 and OPRD1, have been tested, for example, in the European Pharmacogenetic Opioid Study (EPOS). EPOS is the largest genetic association study of opioid response to date, with 2294 patients taking opioids for cancer-related pain. A total of 112 SNPs in 25 genes, including OPRM1, OPRK1 and OPRD1, were investigated for relationship to oral equivalent morphine dose requirements. However, no association was identified with any of the SNPs tested in both development and validation analyses. 57
When morphine response phenotypes were mathematically determined by principal component analysis in one cancer-related pain study (n = 207), two main components were identified: analgesia and central side effects. Multivariate regression analysis was used to combine clinical and genetic factors (ORPM1, OPRD1 and OPRK1 SNPs) to predict response. A total of 16% of variability in analgesic response was predicted by a model, including the OPRK1 SNP rs7824175, two types of concomitant medication: beta-blockers, and anti-emetic and daily morphine dose. A total of 10% of variability in central side effects for morphine was predicted by two SNPs, OPRM1 rs2075572 and OPRK1 rs10504151, including concomitant use of steroid medications, and a diagnosis of sarcoma malignancy. This is an innovative way of defining phenotypes and involving both clinical and genetic factors 58 (Table 1).
Selected post-operative and chronic pain studies assessing polymorphisms in opioid receptor genes and opioid response.
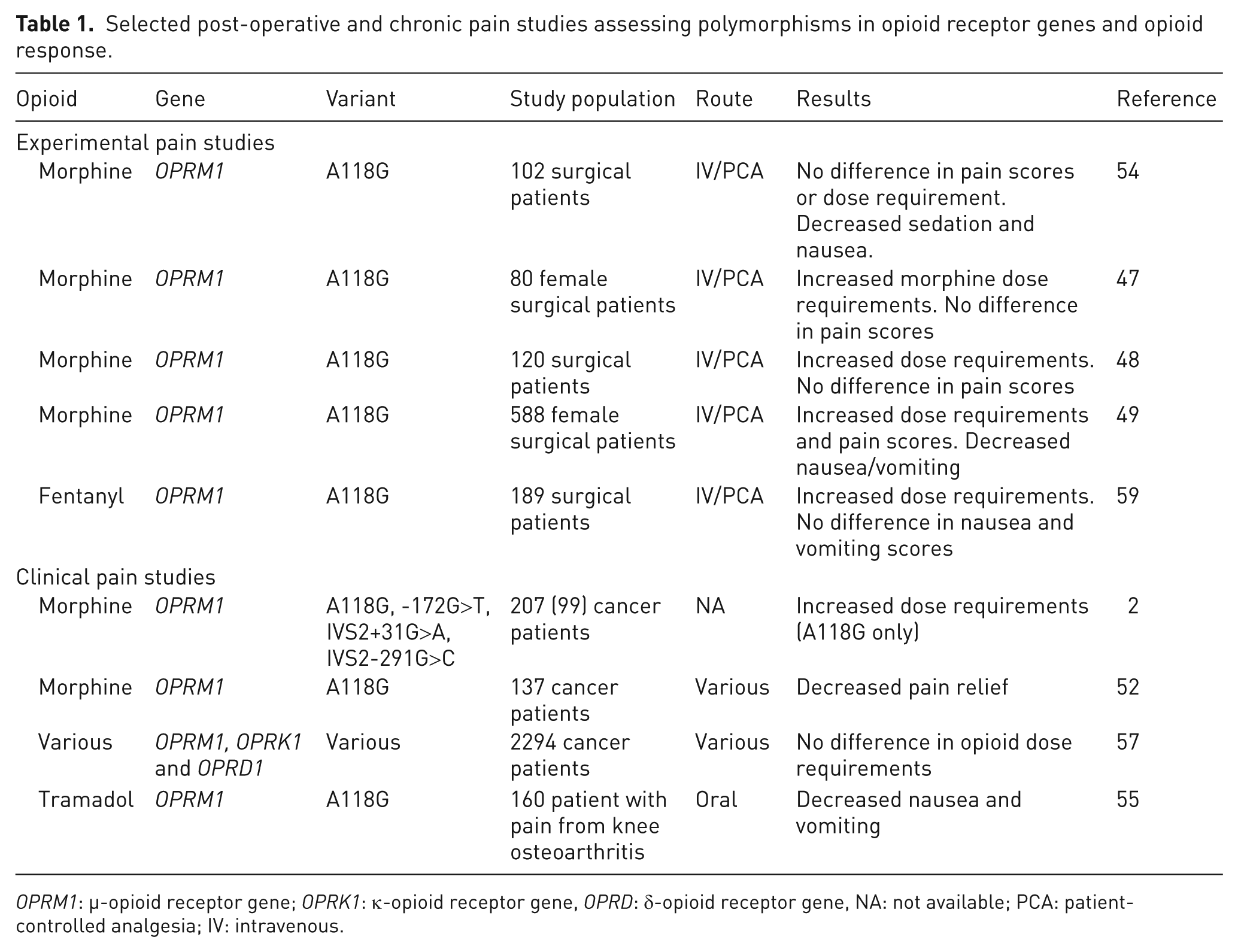
OPRM1: µ-opioid receptor gene; OPRK1: κ-opioid receptor gene, OPRD: δ-opioid receptor gene, NA: not available; PCA: patient-controlled analgesia; IV: intravenous.
STAT6
STAT6 is an important transcription factor involved in the regulation of OPRM1 expression by TH2 cytokines such as interleukin 4 (IL-4). 59 Polymorphisms in STAT6 have been associated with overall response to morphine in cancer-related pain and opioid switching. 60
β-arrestin 2
β-arrestin 2 is an intracellular protein that is integral to µ-opioid receptor inactivation and internalisation. 61 On binding, opioid receptor agonists differentially trigger receptor phosphorylation and recruitment of β-arrestin 2. 62,63 Knockout studies have shown that mice lacking the β-arrestin 2 (ARRB2) gene exhibit prolonged analgesia from morphine treatment at lower doses. 64 It is worth noting, however, that prolonged analgesia in mice lacking β-arrestin 2 may also be due to a combination of more complex effects transduced by multiple GPCRs in the knockout animal model. Polymorphisms in ARRB2 have been associated with overall response to morphine and opioid switching. 60
Pharmacokinetics
Opioid metabolism
Different enzymes in phase 1 and/or phase 2 metabolism are important for the metabolism of different opioids (Figure 2).
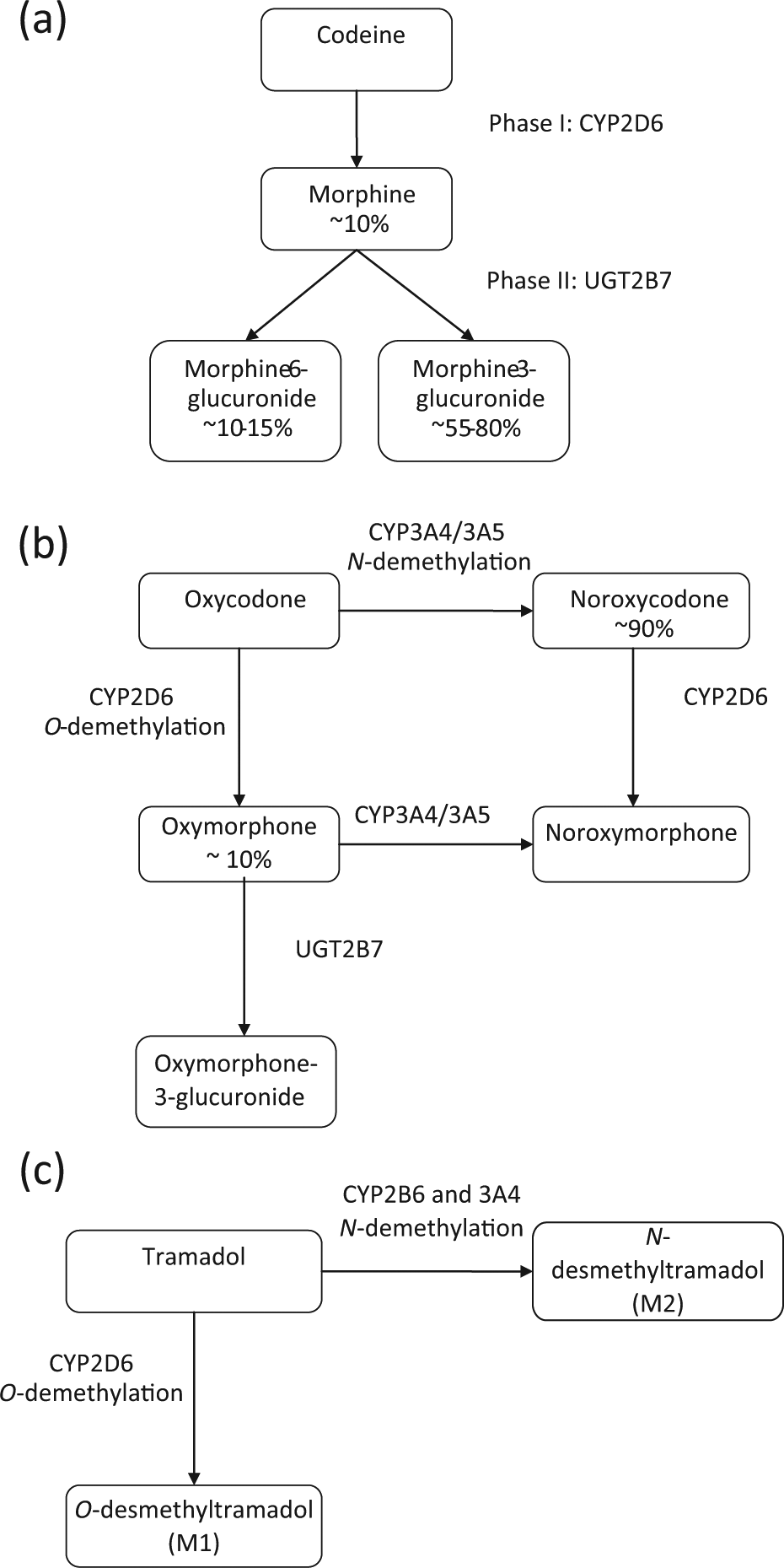
Major metabolic pathways for (a) codeine and morphine, (b) oxycodone and (c) tramadol.
Phase 1 metabolism
Cytochrome P450 2D6
The cytochrome P450 enzyme 2D6 (CYP2D6) is central to the metabolism of several different opioids, including codeine, tramadol and oxycodone, all of which have active metabolites. Over 70 CYP2D6 alleles have been described which have the potential to alter enzyme function, including SNPs, deletions, insertions and copy CNVs. 65 The overall phenotype produced has been classified into four major groups based on function: poor metabolisers, intermediate metabolisers, extensive metabolisers and ultra-rapid metabolisers, for which, tests are available commercially. In Caucasians, approximately 10% of the population are poor metabolisers and 3% are ultra-rapid metabolisers. 66 A small but significant proportion of codeine (10%) is metabolised to morphine by CYP2D6. 67 In response to codeine treatment, poor metabolisers experience little analgesia 68,69 and ultra-rapid metabolisers have a higher incidence of side effects. 1 In addition, there have been cases reported of fatal neonatal opioid toxicity in children breastfed by CYP2D6 ultra-rapid metabolising mothers taking codeine. 70
Oxycodone has two main metabolites: noroxycodone (CYP3A4/5) and oxymorphone (CYP2D6), which account for approximately 90% and 10% of metabolites, respectively. Oxymorphone is reported to have greater analgesic potency compared to oxycodone, whereas noroxycodone is inactive. 71,72 Oxymorphone subsequently is rapidly O-glucuronidated to form oxymorphone-3-β-glucuronide by uridine 5′-diphospho-glucuronosyltransferase-2B7 (UGT2B7) and is excreted so its overall analgesic contribution is probably minimal.
It is currently unclear whether variation in CYP2D6 activity significantly alters the efficacy of oxycodone; experimental studies showing a relationship have not been replicated in the clinical setting. 73 –74 In experimental pain, it has been demonstrated that ultra-rapid metabolisers experience a 1.5- to 6-fold increase in the analgesic effects of oxycodone as compared with extensive metabolisers, and poor metabolisers had a 2- to 20-fold reduction of the analgesic effects as compared to extensive metabolisers. 74 However, a large study of patients receiving oxycodone for cancer-related pain (n = 450) showed that, although CYP2D6 metaboliser status influenced oxycodone metabolite ratios as expected, there was no clinically measurable difference in terms of analgesia or side effects (nausea or sedation). 76
Two post-operative pain studies have found that poor metabolisers use more tramadol when given as patient-controlled analgesia compared to other phenotypes (extensive metabolisers or intermediate metabolisers). 77,78 CYP2D6 metaboliser status has also been linked to tramadol-related side effects, specifically nausea/vomiting. In Korean patients, taking tramadol for osteoarthritis of the knee, CYP2D6 intermediate metabolisers experienced less nausea/vomiting than extensive metabolisers 55 (Table 2).
Selected clinical pain studies assessing polymorphisms in CYP2D6 and opioid response.
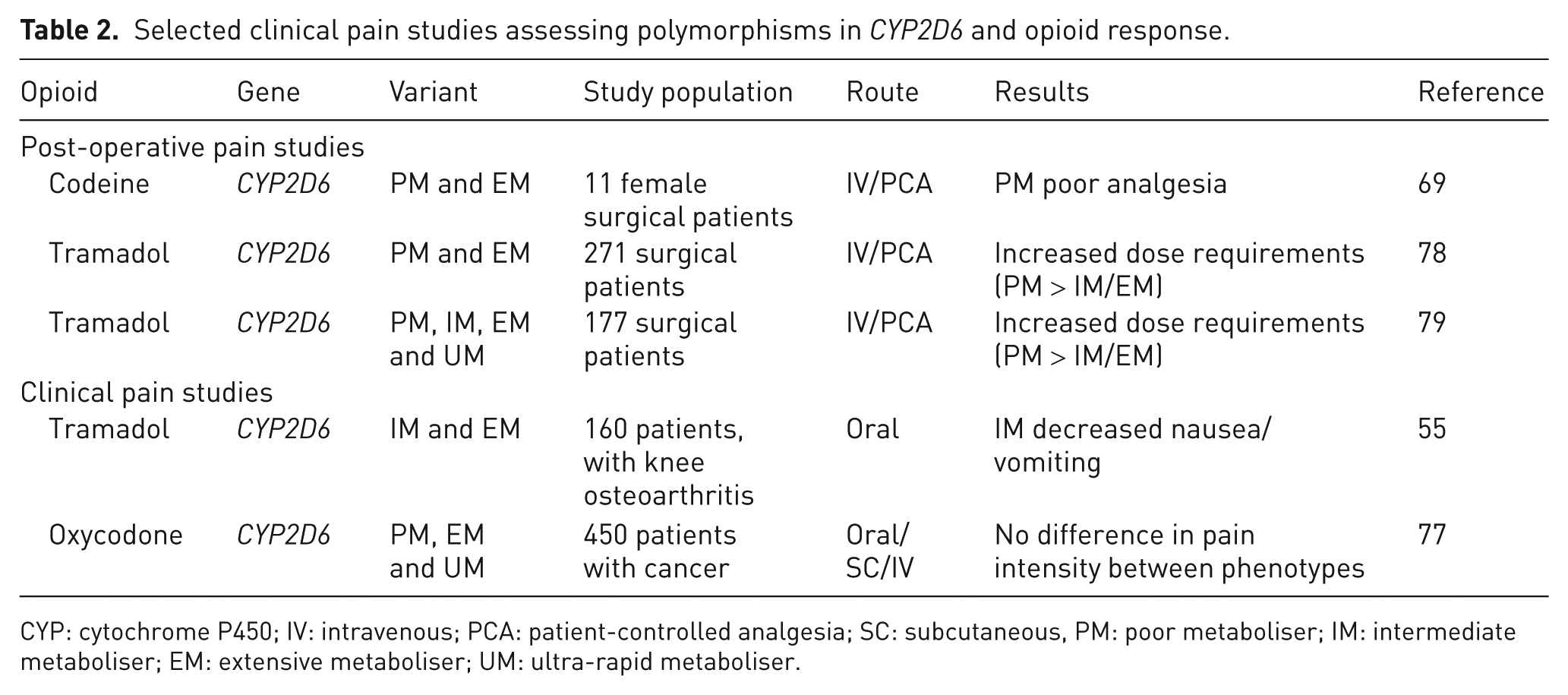
CYP: cytochrome P450; IV: intravenous; PCA: patient-controlled analgesia; SC: subcutaneous, PM: poor metaboliser; IM: intermediate metaboliser; EM: extensive metaboliser; UM: ultra-rapid metaboliser.
Cytochrome P450 3A
The CYP450 3A superfamily of enzymes is involved in the metabolism of 50% of all known drugs. Some 3A substrates, including opioids, for example, oxycodone and fentanyl, can be metabolised equally by 3A4 or 3A5; therefore, a defect in one enzyme may be compensated for by the other. The interaction between 3A4 and 3A5 genetic polymorphisms was studied in Chinese women with post-operative pain following gynaecological surgery. Results showed that while 3A5 variation was not independently important, interactions between 3A4 and 3A5 polymorphisms were additive and significant. 79
Phase 2 metabolism
UGT2B7
The hepatic isoenzyme UGT2B7 is primarily responsible for morphine metabolism. In vitro work has suggested that functional genetic variants in UGT2B7 are linked to altered levels of mRNA expression 80,81 and enzyme activity with differential metabolite production. 81 The main metabolites of morphine: morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) account for approximately 50% and 10% of metabolites, respectively. 82 M3G binds poorly to opioid receptors and may be responsible for neuroexcitatory effects such as hyperalgesia, allodynia and myoclonic jerks. 83 M6G, however, has been used as an analgesic agent in its own right. 84 Clinical studies have linked genetic variation to differences in morphine/metabolite ratios, 85 but not to overall clinical response to morphine. 86
Multi-drug resistance gene
The multi-drug resistance gene or adenosine triphosphate (ATP)-binding cassette subfamily B, member 1 (MDR1 or) encodes P-glycoprotein. P-glycoprotein is a membrane transporter with a central role in the regulation of drugs crossing the blood–brain barrier, and actively removes drugs from the central nervous system (CNS). Heterozygosity for the ABCB1 3435T allele has been associated with decreased morphine equivalent daily dose in a mixed chronic pain population 51 and increased pain relief from morphine in cancer-related pain. 52 Side effects have also been associated with ABCB1 polymorphisms with conflicting results. In an experimental pain study, the variant alleles 2677A and 3453T were protective against nausea and vomiting. 87 However, in a post-operative pain study, use of anti-emetics for morphine-related nausea and vomiting was decreased in patients who were homozygous for the 2677GG/3435CC diplotype. 88 The presence of the A allele at position 2677 of ABCB1 has been reported to be protective of central side effects, that is, drowsiness and confusion, in patients treated with morphine for cancer-related pain. 89 Functional variants changing transporter activity may influence drug concentrations and parent drug/metabolite ratios in the CNS and consequently adverse reactions; G2677T/A has been shown to be linked to altered expression of P-glycoprotein in vivo 90,91 (Table 3).
Selected pain studies assessing polymorphisms in drug transporter (ABCB1) gene and opioid response.
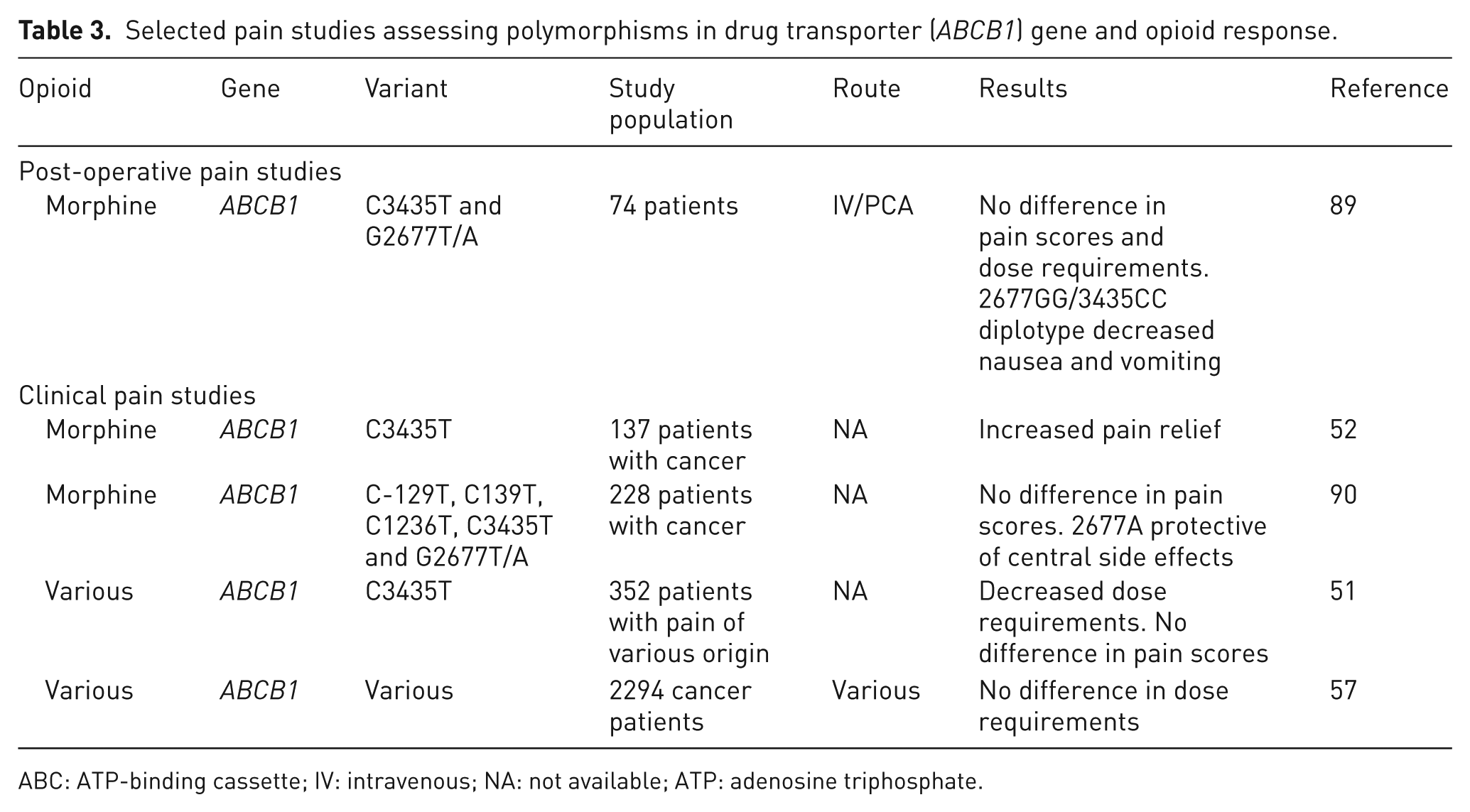
ABC: ATP-binding cassette; IV: intravenous; NA: not available; ATP: adenosine triphosphate.
Modifying systems
Catechol-O-methyltransferase
The enzyme catechol-O-methyltransferase (COMT) metabolises catecholamines, such as noradrenaline and dopamine; therefore, changes in activity may influence neurotransmission. The most commonly studied SNP in the COMT is p.158V>M (rs4680), which results in the substitution of valine to methionine at amino acid position 158. This change has functional consequences as enzyme activity is reduced by between three- and four-fold. The p.158V>M polymorphism has been associated with increased morphine dose requirements in cancer-related pain. 92 Genetic variation in COMT has also been associated with opioid-related side effects in patients treated for cancer-related pain. In a subgroup analysis of EPOS, COMT polymorphism was associated with severity of nausea and vomiting (n = 1579). 93 Three COMT SNPs were found to be weakly associated with less nausea/vomiting: rs165722C, rs4633T and rs4680G, although the significance was lost after correcting for multiple testing. 93 COMT metabolises dopamine, which is an important neurotransmitter in the area postrema and vomiting centre. In cancer patients receiving morphine, the common G allele at position -4873 (rs740603) of COMT has been reported as protective of central side effects. 89 The effect of this allele was independent of and additive to the ABCB1 2677A allele (rs2032582), which demonstrates the importance of considering interactions between multiple genes.
HTR3B
Activation of 5-HT3 (serotonin) receptors in the gastrointestinal tract or chemoreceptor trigger zone is pro-emetic. Three SNPs in the 5-HT receptor 3B gene (HTR3B) have been associated with opioid-related nausea/vomiting in cancer patients in a large study: carriers of rs1176744G, rs3782025T and rs1672717T were found to suffer from less nausea/vomiting. 93 Notably, the association with the G allele of rs1672717 remained significant when corrected for multiple testing.
Cytokines
Cytokines are vital to the co-ordination of the immune system and the inflammatory response. Cytokines may be broadly classified as pro-inflammatory (tumour necrosis factor α (TNFα), IL-6, IL-8) or anti-inflammatory (IL-10, IL-4, transforming growth factor beta β (TGFβ)). Spinal administration of morphine in animal models stimulates the release of pro-inflammatory cytokines by CNS glial cells, and has been shown to inhibit acute opioid analgesia and induce opioid tolerance after repeated administration. 94,95 In cancer-related pain studies, SNPs in several cytokine gene promoters (IL-8, IL-6 and TNF) have been associated with pain severity and morphine dose requirements. 96,97 Polymorphisms in the promoter region may influence transcription factor binding sites, thereby modifying gene expression.
Pharmacogenetics of non-steroidal anti-inflammatory drugs
Non-steroidal anti-inflammatory drugs (NSAIDs) are the most commonly prescribed painkillers in the world, as many of them are easily accessible over the counter; these also possess anti-inflammatory and antipyretic properties. Annual National Health Service (NHS) prescriptions for all causes have reached 25 million in 2012. They are associated with 12,000 hospital admissions per year in order to treat side effects, and they reportedly contribute to 2600 deaths in the United Kingdom per annum. 98,99
Pharmacodynamics
Cyclooxygenase enzymes
The molecular target of NSAIDs is blockade of the cyclooxygenase (COX) enzymes in the arachidonic acid cascade. Inhibition of COX-1 accounts for most of the side effects, 100,101 while COX-2 inhibition produces therapeutic effects. 102,103 Current literature describes variability in the genetic expression of these COX isoforms with functional and sometimes clinically relevant results. 104,105 For instance, carriers of the COX-1 c.1676- >T (rs1330344) allele were found to have a significant risk of non-malignant gastric ulcers when using NSAIDs, 106 while the COX-1 c.50C>T polymorphism was linked to an impaired inhibitory effect on aspirin, 107 although it failed to demonstrate risk of peptic ulcer bleeding. 108
The main COX-2 functional polymorphism is c.765G>C and is associated with a reduced risk of myocardial infarction and stroke, 109 and of developing Crohn’s disease. 110 However, adverse effects have also been identified and include increased monocyte prostaglandin production causing a more severe course of asthmatic disease, reflected by the need for oral corticotherapy, 111 and a significant association with poor outcome in stroke patients from its effect on aspirin resistance. 112
Pharmacokinetics
CYP2C9 is one of the most abundant P450 cytochromes in the liver and works in the phase 1 metabolism of approximately 15% of clinically useful drugs, including various NSAIDs. 113,114
Common polymorphisms of the CYP2C9 gene exist, with three main alleles: CYP2C9*1, CYP2C9*2 and CYP2C9*3. 115 These have been shown to affect cytochrome activity. For example, the allele CYP2C9*3, in which isoleucine 359 is changed to leucine (p.359I>L), shows a marked decrease in CYP2C9 activity, and individuals carrying the homozygous genotype CYP2C9*3/*3 were shown to have between 5- and 10-fold reduced activity depending on the study design. 115 –117 The allelic variants CYP2C9*2, CYP2C9*1/*2 and CYP2C9*1/*3 were also associated with a slower metabolism in a number of drug substrates with up to 50% reduction in the Vmax/Km ratio. 118,119 NSAID pharmacological activity is almost exclusive to the S(−) enantiomer, 120 and CYP2C9 contributes to its metabolism. 121 Studies indicate that NSAID-induced common adverse reactions are probably related to inherited impairment of the CYP2C9 genotype activity. 122 Individuals carrying the gene variants CYP2C8*3 (rs11572080; rs10509681), CYP2C9*2 (rs1799853) or CYP2C9*3 (rs1057910) show an increased risk of developing acute gastrointestinal bleeding following the use of NSAIDs. 123 Similar research revealed that the highest bleeding risk from NSAID use was in patients who possessed both the CYP2C8*3 and CYP2C9*2 alleles. 122 While CYP2C9 contributes to the metabolism of most NSAIDs, recent data show that CYP2C8 polymorphisms may influence inter-individual variability in the pharmacokinetics of some NSAIDs, namely, ibuprofen and diclofenac. 125 Individuals who are homozygous or double-heterozygous for CYP2C8*3 and CYP2C9*3 variant alleles (8% of the population) had extremely low ibuprofen clearance rates, with values ranging from 7% to 27% of the mean clearance rates among non-carriers of mutations. 125
Voltage-gated sodium channel modulators
Voltage-gated sodium channels (VGSCs) expressed at the terminals of nociceptive poly-sensors act as downstream targets in the process of stimulation, and their activation leads to the initiation of action potentials that are propagated from the periphery to the CNS. 126 The VGSC family consists of nine proteins (Nav1.1–Nav1.9) that are expressed on the membrane of excitable cells and allow intermittent passage of Na+ ions into these cells. Three isoforms – Nav1.7, Nav1.8 and Nav1.9 – are predominantly expressed in the peripheral nervous systems, both somatic as well as autonomic. 127 Nav1.7 is thought to serve a ‘threshold channel’ function, so when activated, Nav1.7 (and Nav1.9 in some cells) is likely to bring the neuron towards the threshold, and Nav1.8 is largely responsible for the overshooting action potential. 128,129 In addition to classic local anaesthetic (LA) molecules, sodium channels seem to be modulated by a range of other heterogeneous drugs such as carbamazepine, mexiletine, amitriptyline, ketamine and alcohol, all used as analgesics in clinical practice. These modulators interact with the channel on the molecular level in many different ways. 130
Pharmacodynamics
Mutations within the family of sodium channel genes are known to correlate with varied binding characteristics and clinical actions of the LAs. An example of inherited drug-resistance is demonstrated by the p.395N>K mutation in the SCN9A gene, which produces an increased resistance to lidocaine. 131 Inherited conditions, altering electrogenesis by prolonging fast inactivation of VGSCs, such as PEPD, are known to be preferentially responsive to carbamazepine. 34
SNPs causing the development of inherited erythromelalgia (PEM), leading to the enhancement of channel activation, can be better controlled with oral mexiletine. 132 Treatment of both of these gain-of function sodium channelopathies (PEM and PEPD) with systemic non-selective VGSC blockers has been proven to be rather disappointing overall. 133
In addition to the involvement of MC1R in pain modulation, 134 it appears that red-haired individuals were less affected by the anaesthetic effect of subcutaneous lidocaine, as measured by the pain perception and tolerance thresholds. 135 The MC1R gene is not known to be expressed at the periphery, around the nerve fibres, 136 and it is unclear what association there is between the sodium channel blockade in the periphery and melanocortin-1 G-protein–coupled receptor gene. Interaction at the higher, pain modulatory level could play a role in this mechanism. 137 It remains to be studied how other more subtle variations in pain perception phenotypes, such as the recently described association between the A allele of p.1150R>W (rs6746030) and altered pain threshold, 138 are affected by pharmacological modulation of the sodium channel.
Pharmacokinetics
Ester and amide LAs undergo quite different metabolic processes in humans. Most esters are broken down rapidly by plasma esterases to inactive compounds and are excreted renally. Similar to diamorphine, cocaine breakdown is catalysed by pseudocholinesterases human liver carboxylesterase (hCE) 1 and 2. 140,140 It is well known that polymorphism in the pseudocholinesterase peptide is related with apnoea following administration of the muscle relaxant succinylcholine. 141 What is less known is the fact that individuals who carry an inactive copy of this enzyme are also unable to hydrolyse diamorphine to its active metabolite, morphine, and those who have only partially active pseudocholinesterase do so to a much lesser extent than carriers of a fully active isoform. 142 Xie et al. 143 in 1999 examined how variants of human cholinesterase affect cocaine breakdown and found that the substitution of aspartic acid with glycine at position 70 of the enzyme led to a 10-fold lower binding efficiency for cocaine and 10-fold lower catalytic efficiency. It has long been proposed that individuals who demonstrate this abnormality should wear a medical alert bracelet or a similar identifier in order to highlight the risk of death or permanent damage when exposed to ester-type LAs. 144 While a small amount of ester-type LAs may be administered to atypical homozygotes, as a general rule, these compounds are best avoided. Amide-type LAs should be used instead.
Amide LAs are metabolised by the phase 1 modification process of hydrolysis (by amidases) and oxidation (by CYP450 oxidase system) in the liver. Both of these pathways are slower than plasma ester hydrolysis, so these molecules have a higher tendency to accumulate in human circulation. The authors are not aware of any evidence linking polymorphisms in the genes coding for amidase and LA metabolism, unlike in the cytochrome P450 system, where variations in drug metabolism can occur in up to 30% of people in certain ethnic groups with up to 30-fold magnitude of difference. 145 The main CYP isoforms involved in the oxidation of LAs are CYP3A4 for lidocaine and bupivacaine and CYP1A2 in case of ropivacaine. Activity assays of CYP3A4 reveal 10- to 100-fold inter-individual differences. 146 One C>T SNP in particular, located in intron 6 of the CYP3A4 gene (rs35599367 or CYP3A4*22 variant), was found to significantly affect the metabolism of xenobiotics, which depend on this enzyme. 147 The cytochrome P450 enzyme CYP1A2 metabolises 5–10% of medications in clinical consumption, including ropivacaine and paracetamol. 148 As with any other enzymatic process, there is considerable variation in 1A2 metabolic activity primarily due to three variables: genetic factors, environmental factors and drug–drug interference. The wild-type allele is conventionally labelled as CYP1A2*1. Two functional SNPs have been identified in this gene. G3860A (CYP1A2*1C type) is associated with decreased metabolic activity in the enzyme produced when compared with the control carriers. The CYP1A2*1F allele is the result of a single point mutation (C163A) and is linked to a hypermetabolic phenotype, particularly under the influence of environmental nicotine when compared with the CYP1A2*1A variant. 149,150 (Table 4). Carriers of the 1A2 hyperinduction phenotype, mostly those of Japanese, Egyptian or Caucasian origin, account for up to 45% of the population. 149,151 Commercially available kits testing for these 1A2 variants using polymerase chain reaction (PCR), allele-specific primer extension and subsequent hybridisation using immobilised nucleic acid probes enabling fluorescent detection already exist.
Selected experimental and chronic pain studies assessing polymorphisms in genes involved in action of sodium channel blockers.
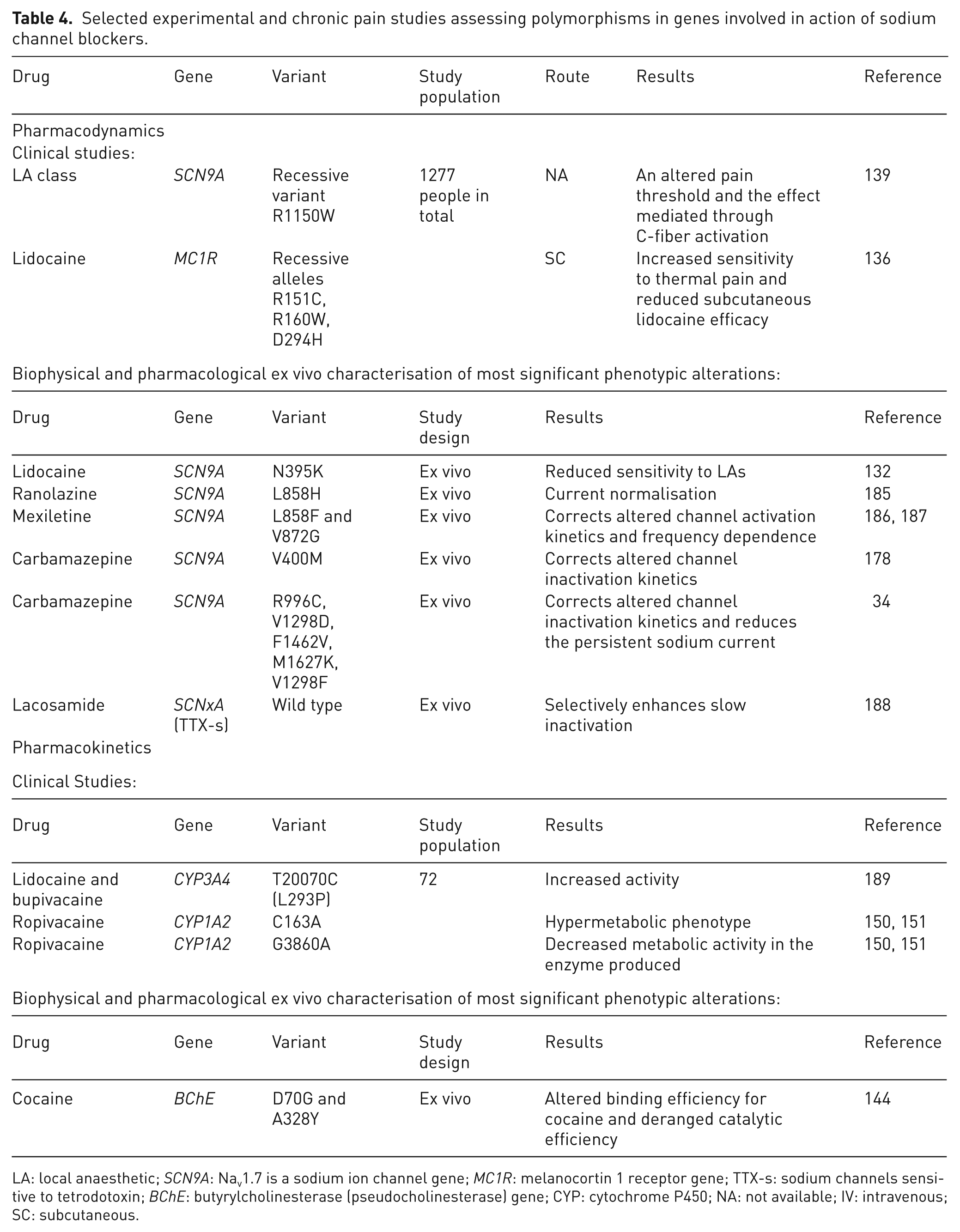
LA: local anaesthetic; SCN9A: Nav1.7 is a sodium ion channel gene; MC1R: melanocortin 1 receptor gene; TTX-s: sodium channels sensitive to tetrodotoxin; BChE: butyrylcholinesterase (pseudocholinesterase) gene; CYP: cytochrome P450; NA: not available; IV: intravenous; SC: subcutaneous.
Other analgesics
Paracetamol
Paracetamol (N-(4-hydroxyphenyl)-acetamide) is one of the most widely used over-the-counter analgesics. Many studies indicate that paracetamol can offer rapid pain relief for acute pain. 152,153 In chronic pain, paracetamol can be effectively used in treating migraine pain 154 and osteoarthritis. 155 The pharmacodynamic properties of paracetamol, as well as the newly described active metabolite AM404 molecule, 156 are inadequately explained and may involve a diverse range of pathways which include transient receptor potential cation channel subfamily V member 1 (TRPV1) receptors via inhibition of the reuptake of the endogenous cannabinoid/vanilloid anandamide, modulation of VGSC currents, 157 5-HT receptors 158 or the COX system. 159 Candidate genes involved in these biological systems, with time, are likely to reveal a degree of inter-individual differences.
Paracetamol hepatotoxicity is the most common cause of acute liver failure in the United Kingdom. 160 Under normal conditions, paracetamol is extensively conjugated with glucuronic acid and sulphate as part of phase 2 metabolism in order to make it water soluble, preceding its excretion via the kidneys. A total of 5% of the remaining drug undergoes phase 1 oxidation in the liver via the CYP system. Cytochrome P450 2E1 and 3A4 convert paracetamol to a toxic intermediary metabolite, N-acetyl-p-benzoquinoneimine (NAPQI), which is instantly cleared by conjugation with glutathione to form cysteine and other conjugates. 161 This glucuronidation process was first noted to be impaired in sufferers from the inherited bilirubin disglucuronidation condition called Gilbert’s syndrome, increasing the risk of paracetamol toxicity in affected individuals. 162 Furthermore, evidence, collected by Patel et al. 163 indicated that up to 33% of Oriental subjects displayed relatively extensive glucuronidation with clinically relevant lower incidence of a fulminant liver failure in patients belonging to this ethnic group who ingested large amounts of paracetamol. 164 Activity of CYP2E1 can be decreased by variety of environmental factors such as liver cirrhosis, chronic alcohol abuse and so on. 165
Ketamine
Ketamine is metabolised to several phase 1 metabolites, including alkylhydroxy-ketamine, nor-ketamine and dihydro-norketamine. CYP enzymes involved in this process are 3A4 (>60% metabolism), 2C9 and 2B6. 166 Norketamine subsequently undergoes phase I liver processing with the aid of 2B6 and 2A6. 167 When tested in a Swedish Caucasian population, 3A4 normal and slow metabolisers demonstrated no difference in overall pharmacokinetic parameters or in ketamine-related side effects. 168
Tricyclic antidepressants
Amitriptyline belongs to the tricyclic antidepressant (TCA) group of drugs. It has been a first-line treatment for neuropathic pain and fibromyalgia for many years. 169,170 Disappointingly, however, there is still little robust evidence for a beneficial outcome in treating of these chronic pain states. 171 Amitriptyline acts as a combined serotonin–norepinephrine reuptake inhibitor as well as a sodium channel blocker. Descending noradrenergic inhibitory mechanisms are augmented by this class of drugs, and this is thought to be the main mechanism of the anti-neuropathic action of amitriptyline. 172 The norepinephrine transporter (NET) is a peptide that is encoded by the SLC6A2 gene. SLC6A2 polymorphism seems to be associated with altered pain thresholds in humans. 173 The influence of SNPs in SLC6A2 on the efficacy and TCAs in patients suffering with neuropathic pain has not yet been studied. 174
Carbamazepine
An anticonvulsant carbamazepine is used in post-herpetic and trigeminal neuralgias as well as autoimmune-mediated pain states such as Guillain–Barré syndrome. 175 Rare heritable severe pain conditions such as PEPD and some forms of inherited erythromelalgia are treated with this drug as well. 176 Characterisation of sodium current alterations caused by p.1627M>K, p.1464T>I mutations affecting SCN9A gene have revealed that the likely mechanism of action of carbamazepine is via a normalisation of voltage dependence of inactivation and activation in VGSC action. 177 –179
Carbamazepine is mainly metabolised by the CYP3A4 enzyme to carbamazepine-10,11-epoxide. This drug has been linked to severe, type B (idiosyncratic, dose-independent) adverse cutaneous and systemic reactions varying from Stevens–Johnson syndrome (SJS) to toxic epidermal necrolysis (TEN). 180,181 There is an association of the development of both TEN and SJS in carriers of the human leukocyte antigen (HLA) HLA-B*1502 allele. This is observed explicitly in Asians who are prescribed carbamazepine. More recently, both HLA*3101 and HLA*1511 alleles have also been identified as potentially contributory to the increased risk of development of these reactions. 182 –184 As a result, the Food and Drug Administration (FDA) recommends that before introducing carbamazepine, all Asian patients be genotyped for the HLA-B*1502 allele.
Discussion
Pharmacogenetics has the potential to provide clinical guidance on drug dosing and timing in order to reach maximum efficacy and minimum side effects and complications. However, with the vast scope of genetic variables likely to contribute to pain phenotypes, ‘bedside’ clinically available kits have limited applicability. 185
There have been two main approaches to population-based genetic association studies: candidate gene studies and, more recently, genome-wide association (GWA) studies. Candidate gene studies tend to focus on a small set of SNPs in genes, which are hypothesised to have biological relevance to the condition being studied. In analgesic response studies, these have mainly been in key genes from either pharmacodynamic or pharmacokinetic pathways. The SNPs selected usually include functional SNPs, which may have direct causal relevance. GWA arrays can type as many as one million SNPs across the genome to provide the highest possible coverage of common genetic variation. Associations generated from GWA studies may not have any direct causal relevance and are more likely to be in linkage disequilibrium with underlying causative variants. This approach may also identify novel contributing genes previously unidentified in our current understanding of pain pathways and represents an exciting technique for future investigations.
Population-based genetic association studies, which aim to correlate genotype to phenotype in complex traits, including pain and analgesic response, have had variable success; the reproducibility of results has remained low. Twin studies demonstrate that up to 60% of the observed variability in response to painful stimuli is genetically determined. However, genetic and environmental factors known to contribute to pain experience are only moderately correlated across different pain modalities, which suggests that different genes influence different types of pain. In pain of mixed aetiology, such as cancer-related pain, genetic influences may therefore not be clearly identified in clinical studies. 186
The majority of genetic association studies that investigate inter-individual variability in analgesic response have used relatively small sample sizes. There are several factors which contribute to the required sample size, including the prevalence of disease/trait in the general population, the frequency of the susceptibility allele and its effect size and the number of SNPs to be tested. The lower the frequency of the susceptibility allele and lower the effect size, the larger the sample size required. Complex traits are likely to be influenced by multiple genetic variables, all with small or modest effect sizes. Any variant strongly associated with a disease or trait is likely to be rare. 187 Therefore, large sample sizes, possibly of many thousands, are generally preferable in the study of complex traits. The sample sizes in the studies described in this narrative review are generally small and therefore many associations, particularly with small effect sizes, may not have been identified.
The candidate gene approach used to study rare dramatic human phenotypes has identified a variety of promising therapeutic targets. NTRK1, SCN9A and P2X family of genes have been the focus of drug development for the last decade with some molecules reaching phase 3 clinical trials. Undesirable side effects and idiosyncratic reactions aside, as a proof of concept, these examples greatly encourage more work and research to be done in order to identify more potential drug targets. A recent review article produced by Lotsch and Geisslinger 188 has explored this particular area further.
Pain experience and analgesic response are complex traits, and as such are likely to be influenced by a host of gene–gene and gene–environment interactions. A few studies have started to investigate interactions between polymorphisms from more than one gene; however, so far, this has been limited to two candidate SNPs at once. 90,189 Environmental and patient variables such as compliance, concomitant medications, diet and psychosocial issues also contribute to the ultimate endpoint of analgesic response. The exploration of potential gene–gene/gene–environment interactions or epistasis provides a huge challenge for future pharmacogenomic research, both practical and analytical. Such work requires exponential increases in sample size and focused phenotype definitions.
Other variations besides the DNA sequence may influence phenotype in epigenetic processes, for example, histone modifications and DNA methylation. The purpose of this complex process seems to be the activation or silencing of specific genes. The inherited phenotypic change may be achieved without any alteration in the DNA sequence. 190,191 This phenomenon has already been associated with many other neural functions, including plasticity of synaptic transmission and memory. In the context of peripheral nerve injury, animal studies have revealed epigenetic changes, down-regulating the expression of some members of both the opioid and sodium channel expressing family of genes. 192 This is achieved via the neuron-restrictive silencer factor (NSRF), which is a transcriptional repressor of genes expressed in the peripheral C-fibres. Nociceptor-related targets include OPRM1, SCN10A and KCND3. 193 It remains to be seen if coding polymorphisms of the NSRF complex and related neuron-restrictive silencer element (NRSE) in any way influence our pain perception and alter the way analgesic drugs interact with humans.
The majority of the human genome is transcribed into non-protein coding RNA (ncRNA) molecules, which seem to play a part in genetic adjustments of our traits. 194 Other molecules, 20- to 30-base-pair-long RNAs called microRNAs (miRNAs), target specific sites and alter phenotypes by regulating expression of the whole cluster of genes in a tissue-specific manner. We have only just started to appreciate their influential role on nociceptor-related gene expression patterns and regulation of pain signalling. 195 How to utilise this knowledge of miRNA represents an exciting new chapter of drug discovery.
Any further implementation of pharmacogenetic assays into day-to-day pain management practice faces many obstacles such as ethical, legal and social issues, a lack of readily available resources, as well as the scientific quality of information itself. 196
Conclusion
Phenotypic differences in pain perception and its pharmacological modulation are significantly dependent on human genetic factors. Knowledge about genes governing pharmacodynamic and pharmacokinetic processes involving analgesic molecules is gaining more consideration among prescribers. Lack of robustness and reproducibility in pain pharmacogenetics correlation studies is one of many significant limitations in development of readily available bedside genotyping devices. Personalised drug selection and dosing for individual patients with acute or chronic pain is still a long way off.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.